REFINEMENT. PROGRAM : GSAS AUTHORS : LARSON & VON DREELE DATA USED IN REFINEMENT RESOLUTION RANGE ... REFINEMENT. PROGRAM : GSAS AUTHORS : LARSON & VON DREELE DATA USED IN REFINEMENT RESOLUTION RANGE HIGH (ANGSTROMS) : 2.27 RESOLUTION RANGE LOW (ANGSTROMS) : 15.29 POWDER DIFFRACTION DATA. FIT TO DATA USED IN REFINEMENT NUMBER OF POWDER PATTERNS : 4 PROFILE R VALUES (%) : 2.97 2.90 2.91 3.28 WEIGHTED PROFILE R VALUES (%) : 4.24 4.02 3.94 4.44 F**2 R VALUES (%) : 21.78 24.94 28.10 14.50 NUMBER OF POWDER PATTERN POINTS : 9100 10666 6154 4359 NUMBER OF REFLECTIONS : 3317 3348 1865 1719 TOTAL NUMBER OF POWDER POINTS : 30279 NUMBER OF NON-HYDROGEN ATOMS USED IN REFINEMENT. PROTEIN ATOMS : 544 NUCLEIC ACID ATOMS : NULL HETEROGEN ATOMS : NULL SOLVENT ATOMS : 36 MODEL REFINEMENT. NUMBER OF LEAST-SQUARES PARAMETERS : 1752 NUMBER OF RESTRAINTS : 1980 LEAST-SQUARES MATRIX BAND WIDTH : 5 MARQUARDT COEFFICIENT : 5.86 RMS DEVIATIONS FROM RESTRAINT TARGET VALUES. NUMBER. BOND ANGLES (DEG) : 2.49 740 INTERATOMIC DISTANCES (A) :0.010 556 TORSION ANGLE RESTRAINTS (E) : 0.30 205 CHIRAL VOLUMES (A**3) :0.161 72 DISTANCES FROM RESTRAINT PLANES (A) :0.014 439 TORSION PSEUDOPOTENTIAL RESTRAINTS (E) : 2.34 92 ANTI-BUMPING DISTANCE RESTRAINTS (A) :0.025 102 HYDROGEN BOND DISTANCE RESTRAINTS (A) :0.170 77 EXPERIMENTAL DETAILS EXPERIMENT TYPE : X-RAY POWDER DIFFRACTION DATE OF DATA COLLECTION : 13-MAR-2006 TEMPERATURE (KELVIN) : 295 PH : SAMPLE HOLDER : 1.5MM GLASS CAPILLARY NUMBER OF CRYSTALS USED : POLYCRYSTAL SLURRY SYNCHROTRON (Y/N) : Y RADIATION SOURCE : ESRF BEAMLINE : ID31 X-RAY GENERATOR MODEL : NULL MONOCHROMATIC OR LAUE (M/L) : M WAVELENGTH OR RANGE (A) : 0.8012034(76), 1.251209(40), 1.242481(32) MONOCHROMATOR : DOUBLE SI(111) OPTICS : 2MM X 8MM BEAM ANALYZER : SI(111) DETECTOR TYPE : NINE AVALANCHE PHOTODIODE (APD) DETECTOR MANUFACTURER : NULL INTENSITY-INTEGRATION SOFTWARE : NULL DATA SCALING SOFTWARE : ID31SUM SOFTWARE USED: GSAS STARTING MODEL: PDB ENTRY 1W70
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) SYNCHROTRON / Resolution: 2.27 Å
SYNCHROTRON / Resolution: 2.27 Å  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads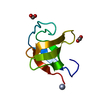








 PDBj
PDBj



 Assembly
Assembly

 Mass: 18.015 Da / Num. of mol.: 36 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 36 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID13 / Wavelength: 0.8012034, 1.241209, 1.242481
/ Beamline: ID13 / Wavelength: 0.8012034, 1.241209, 1.242481 Processing
Processing