 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2mcm | ||||||
|---|---|---|---|---|---|---|---|
| Title | MACROMOMYCIN | ||||||
 Components Components | MACROMOMYCIN | ||||||
 Keywords Keywords | APOPROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Streptomyces macromomyceticus (bacteria) Streptomyces macromomyceticus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.5 Å X-RAY DIFFRACTION / Resolution: 1.5 Å | ||||||
 Authors Authors | Van Roey, P. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.Usa / Year: 1989 Title: Crystal structure analysis of auromomycin apoprotein (macromomycin) shows importance of protein side chains to chromophore binding selectivity. Authors: Van Roey, P. / Beerman, T.A. | ||||||
| History |
| ||||||
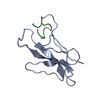
| Remark 700 | SHEET THE STRUCTURE CONSISTS OF A SEVEN-STRANDED ANTIPARALLEL BETA-BARREL (FLATTENED) AND TWO ...SHEET THE STRUCTURE CONSISTS OF A SEVEN-STRANDED ANTIPARALLEL BETA-BARREL (FLATTENED) AND TWO ANTIPARALLEL BETA-RIBBONS. THE FOLDING OF THE BETA-BARREL HAS SIMILAR GREEK KEY MOTIF AS THE C DOMAIN OF IMMUNOGLOBULIN. THE STRUCTURE IS COMPOSED OF A FLATTENED SEVEN-STRANDED ANTIPARALLEL BETA-BARREL AND TWO ANTIPARALLEL BETA-SHEET RIBBONS. THE BARREL AND RIBBONS DEFINE A DEEP CLEFT THAT IS THE CHROMOPHORE BINDING SITE. RESIDUES LISTED UNDER SITE REFER TO RESIDUES THAT HAVE SIDE CHAINS EXTENDING INTO THIS AREA. THE SHEET PRESENTED AS *BRL* ON SHEET RECORDS BELOW IS ACTUALLY A SEVEN-STRANDED BETA-BARREL. THIS IS REPRESENTED BY A EIGHT-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2mcm.cif.gz | 36.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2mcm.ent.gz | 23.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2mcm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mc/2mcmftp://data.pdbj.org/pub/pdb/validation_reports/mc/2mcm | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUE PRO 8 IS A CIS PROLINE. |
-Components
| #1: Protein | Mass: 10755.872 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptomyces macromomyceticus (bacteria)References: UniProt: P01549 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca | ||||||||||
| #3: Chemical |   Mass: 118.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#4: Chemical | ChemComp-MPD / ( |   Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 109 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 109 / Source method: isolated from a natural source / Formula: H2OCompound details | THE CHROMOPHORE BINDING SITE IS LOCATED BETWEEN THE BARREL AND THE RIBBONS. RESIDUES LISTED ON SITE ...THE CHROMOPHOR | Has protein modification | Y | Nonpolymer details | THE CALCIUM COORDINATION IS A DISTORTED PENTAGONAL BIPYRAMID. LIGANDS COME FROM THREE DIFFERENT ...THE CALCIUM COORDINATI | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 45.34 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal | *PLUS Density % sol: 44 % | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 8 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
- Processing
Processing
| Software | Name: PROFFT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.5→10 Å / σ(F): 2 Details: A NUMBER OF WATERS ARE DISORDERED AND ARE PRESENTED IN TWO OR MORE ALTERNATE CONFORMATIONS. BECAUSE OF THE REFINEMENT PROCEDURE USED, THE OCCUPANCIES DO NOT ADD UP TO 1.0 FOR WATERS IN ALTERNATE CONFORMATIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROFFT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.179 / Highest resolution: 1.6 Å / Lowest resolution: 8 Å / Num. reflection obs: 12674 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
