 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2lbd: LIGAND-BINDING DOMAIN OF THE HUMAN RETINOIC ACID RECEPTOR GAMMA B... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2lbd | ||||||
|---|---|---|---|---|---|---|---|
| Title | LIGAND-BINDING DOMAIN OF THE HUMAN RETINOIC ACID RECEPTOR GAMMA BOUND TO ALL-TRANS RETINOIC ACID | ||||||
 Components Components | RETINOIC ACID RECEPTOR GAMMA | ||||||
 Keywords Keywords | NUCLEAR RECEPTOR / RETINOIC ACID RECEPTOR / ALL-TRANS RETINOIC ACID / LIGAND-BINDING DOMAIN / COMPLEX / HOLO FORM / TRANSCRIPTION REGULATION / LIGAND-DEPENDENT / ACTIVE CONFORMATION / Structural Proteomics in Europe / SPINE / Structural Genomics | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of myeloid cell differentiation / Harderian gland development / trachea cartilage development / glandular epithelial cell development / growth plate cartilage chondrocyte growth / embryonic eye morphogenesis / embryonic camera-type eye development / prostate gland epithelium morphogenesis / embryonic hindlimb morphogenesis / negative regulation of chondrocyte differentiation ...regulation of myeloid cell differentiation / Harderian gland development / trachea cartilage development / glandular epithelial cell development / growth plate cartilage chondrocyte growth / embryonic eye morphogenesis / embryonic camera-type eye development / prostate gland epithelium morphogenesis / embryonic hindlimb morphogenesis / negative regulation of chondrocyte differentiation / positive regulation of programmed cell death / anterior/posterior pattern specification / Signaling by Retinoic Acid / regulation of myelination / regulation of cell size / face development / response to retinoic acid / canonical Wnt signaling pathway / nuclear retinoid X receptor binding / negative regulation of stem cell proliferation / retinoic acid receptor signaling pathway / cellular response to retinoic acid / stem cell proliferation / cellular response to leukemia inhibitory factor / neural tube closure / Nuclear Receptor transcription pathway / nuclear receptor activity / multicellular organism growth / RNA polymerase II transcription regulator complex / Activation of anterior HOX genes in hindbrain development during early embryogenesis / sequence-specific double-stranded DNA binding / transcription regulator complex / DNA-binding transcription factor activity, RNA polymerase II-specific / cell differentiation / RNA polymerase II cis-regulatory region sequence-specific DNA binding / positive regulation of apoptotic process / DNA-binding transcription factor activity / negative regulation of cell population proliferation / apoptotic process / positive regulation of cell population proliferation / chromatin binding / positive regulation of gene expression / chromatin / negative regulation of transcription by RNA polymerase II / positive regulation of transcription by RNA polymerase II / DNA binding / zinc ion binding / nucleoplasm / membrane / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SIR / Resolution: 2.06 Å X-RAY DIFFRACTION / SYNCHROTRON / SIR / Resolution: 2.06 Å | ||||||
 Authors Authors | Renaud, J.-P. / Rochel, N. / Ruff, M. / Moras, D. / Structural Proteomics in Europe (SPINE) | ||||||
 Citation Citation | Journal: Nature / Year: 1995 Title: Crystal structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Authors: Renaud, J.P. / Rochel, N. / Ruff, M. / Vivat, V. / Chambon, P. / Gronemeyer, H. / Moras, D. #1: Journal: Nature / Year: 1995Title: Crystal Structure of the Ligand-Binding Domain of the Human Nuclear Receptor Rxr-Alpha Authors: Bourguet, W. / Ruff, M. / Chambon, P. / Gronemeyer, H. / Moras, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2lbd.cif.gz | 77.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2lbd.ent.gz | 58.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2lbd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lb/2lbdftp://data.pdbj.org/pub/pdb/validation_reports/lb/2lbd | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |











-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 30193.055 Da / Num. of mol.: 1 / Fragment: LBD (LIGAND-BINDING DOMAIN), RESIDUES 178 - 423 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human)Description: CDNA CLONING\: KRUST ET AL., PROC.NATL.ACAD.SCI.USA,86,5310-5314,1989 Cell line: BL21 / Cellular location: NUCLEUS / Gene: HUMAN RAR GAMMA A CDNA (NUCLEOTIDES 946 - 1683) / Plasmid: PET-15B / Species (production host): Escherichia coli / Cellular location (production host): CYTOPLASM / Gene (production host): HRARGAMMA / Production host:  |
|---|---|
| #2: Chemical | ChemComp-REA / 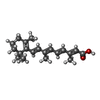  Mass: 300.435 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O2 Mass: 300.435 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O2 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 119 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 119 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.36 Å3/Da / Density % sol: 33 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 7 Details: VAPOR DIFFUSION METHOD, HANGING DROP TECHNIQUE 5UL PROTEIN SOLUTION + 5UL RESERVOIR AGAINST 500UL RESERVOIR, pH 7.0, vapor diffusion - hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 15 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 268 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: LURE  / Beamline: DW32 / Wavelength: 0.901 / Beamline: DW32 / Wavelength: 0.901 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 1, 1994 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.901 Å / Relative weight: 1 |
| Reflection | Resolution: 2.06→7.99 Å / Num. obs: 17193 / % possible obs: 94.08 % / Observed criterion σ(I): 0 / Redundancy: 3.77 % / Rmerge(I) obs: 0.0974 / Net I/σ(I): 22.8 |
| Reflection shell | Resolution: 2.06→2.1 Å / Redundancy: 1.9 % / Rmerge(I) obs: 0.3323 / Mean I/σ(I) obs: 3.64 / % possible all: 57.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIR / Resolution: 2.06→8 Å / Cross valid method: THROUGHOUT / σ(F): 3
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.06→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.21 / Rfactor Rwork: 0.21 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
