positive regulation of glycoprotein metabolic process / growth hormone secretion / COPII-coated vesicle cargo loading / RAB geranylgeranylation / melanosome transport / RAB GEFs exchange GTP for GDP on RABs / Golgi Cisternae Pericentriolar Stack Reorganization / vesicle transport along microtubule / COPII-mediated vesicle transport / COPI-dependent Golgi-to-ER retrograde traffic ...positive regulation of glycoprotein metabolic process / growth hormone secretion / COPII-coated vesicle cargo loading / RAB geranylgeranylation / melanosome transport / RAB GEFs exchange GTP for GDP on RABs / Golgi Cisternae Pericentriolar Stack Reorganization / vesicle transport along microtubule / COPII-mediated vesicle transport / COPI-dependent Golgi-to-ER retrograde traffic / virion assembly / transport vesicle membrane / Golgi organization / endoplasmic reticulum to Golgi vesicle-mediated transport / autophagosome assembly / COPI-mediated anterograde transport / vesicle-mediated transport / endomembrane system / substrate adhesion-dependent cell spreading / positive regulation of interleukin-8 production / small monomeric GTPase / intracellular protein transport / autophagy / endocytosis / melanosome / cell migration / G protein activity / early endosome / defense response to bacterium / cadherin binding / Golgi membrane / GTPase activity / GTP binding / Golgi apparatus / endoplasmic reticulum / extracellular exosome / metal ion binding / cytosol Similarity search - Function
: / : / ARF-like small GTPases; ARF, ADP-ribosylation factor / Small GTPase Rab domain profile. / Ran (Ras-related nuclear proteins) /TC4 subfamily of small GTPases / Rho (Ras homology) subfamily of Ras-like small GTPases / Ras subfamily of RAS small GTPases / Small GTPase / Ras family / Rab subfamily of small GTPases ...: / : / ARF-like small GTPases; ARF, ADP-ribosylation factor / Small GTPase Rab domain profile. / Ran (Ras-related nuclear proteins) /TC4 subfamily of small GTPases / Rho (Ras homology) subfamily of Ras-like small GTPases / Ras subfamily of RAS small GTPases / Small GTPase / Ras family / Rab subfamily of small GTPases / Small GTP-binding protein domain / P-loop containing nucleotide triphosphate hydrolases / Rossmann fold / P-loop containing nucleoside triphosphate hydrolase / 3-Layer(aba) Sandwich / Alpha Beta Similarity search - Domain/homology
BIOMOLECULE: 1 REMARK 300 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT REMARK 300 WHICH ...BIOMOLECULE: 1 REMARK 300 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT REMARK 300 WHICH CONSISTS OF 1 CHAIN(S). THE BIOLOGICAL UNIT OF THIS PROTEIN IS UNKNOWN.
Remark 42
MOLPROBITY STRUCTURE VALIDATION PROGRAMS : MOLPROBITY (KING, REDUCE, AND PROBE) AUTHORS : I.W. ...MOLPROBITY STRUCTURE VALIDATION PROGRAMS : MOLPROBITY (KING, REDUCE, AND PROBE) AUTHORS : I.W.DAVIS,J.M.WORD URL : HTTP://KINEMAGE.BIOCHEM.DUKE.EDU/MOLPROBITY/ AUTHORS : J.S.RICHARDSON,W.B.ARENDALL,D.C.RICHARDSON REFERENCE : NEW TOOLS AND DATA FOR IMPROVING : STRUCTURES, USING ALL-ATOM CONTACTS : METHODS IN ENZYMOLOGY. 2003;374:385-412. MOLPROBITY OUTPUT SCORES: ALL-ATOM CLASHSCORE : 18.17 (0.00 B<40) BAD ROTAMERS : 6.6% 9/137 (TARGET 0-1%) RAMACHANDRAN OUTLIERS : 0.0% 0/151 (TARGET 0.2%) RAMACHANDRAN FAVORED : 95.4% 144/151 (TARGET 98.0%)
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

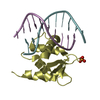









 PDBj
PDBj








 Assembly
Assembly


 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
 Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM
Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM
 Num. of mol.: 2 / Source method: obtained synthetically
Num. of mol.: 2 / Source method: obtained synthetically Mass: 18.015 Da / Num. of mol.: 5 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 5 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing