 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2fda: Crystal Structure of the Catalytic Domain of Human Coagulation Fa... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2fda | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Catalytic Domain of Human Coagulation Factor XIa in Complex with alpha-Ketothiazole Arginine Derived Ligand | ||||||
 Components Components | Coagulation factor XI | ||||||
 Keywords Keywords | HYDROLASE / FXIa / Inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationcoagulation factor XIa / serine-type aminopeptidase activity / Defective F9 activation / positive regulation of fibrinolysis / plasminogen activation / : / serine-type peptidase activity / blood coagulation / heparin binding / serine-type endopeptidase activity ...coagulation factor XIa / serine-type aminopeptidase activity / Defective F9 activation / positive regulation of fibrinolysis / plasminogen activation / : / serine-type peptidase activity / blood coagulation / heparin binding / serine-type endopeptidase activity / : / extracellular exosome / extracellular region / membrane / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 2 Å X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 2 Å | ||||||
 Authors Authors | Jin, L. / Pandey, P. / Babine, R.E. / Weaver, D.T. / Abdel-Meguid, S.S. / Strickler, J.E. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2006 Title: Synthesis, SAR exploration, and X-ray crystal structures of factor XIa inhibitors containing an alpha-ketothiazole arginine Authors: Deng, H. / Bannister, T.D. / Jin, L. / Babine, R.E. / Quinn, J. / Nagafuji, P. / Celatka, C.A. / Lin, J. / Lazarova, T.I. / Rynkiewicz, M.J. / Bibbins, F. / Pandey, P. / Gorga, J. / Meyers, ...Authors: Deng, H. / Bannister, T.D. / Jin, L. / Babine, R.E. / Quinn, J. / Nagafuji, P. / Celatka, C.A. / Lin, J. / Lazarova, T.I. / Rynkiewicz, M.J. / Bibbins, F. / Pandey, P. / Gorga, J. / Meyers, H.V. / Abdel-Meguid, S.S. / Strickler, J.E. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2005Title: Mutation of Surface Residues to Promote Crystallization of Activated Factor Xi as a Complex with Benzamidine: An Essential Step for the Iterative Structure-Based Design of Factor Xi Inhibitors Authors: Jin, L. / Pandey, P. / Babine, R.E. / Weaver, D.T. / Abdel-Meguid, S.S. / Strickler, J.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2fda.cif.gz | 69.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2fda.ent.gz | 50.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2fda.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fd/2fdaftp://data.pdbj.org/pub/pdb/validation_reports/fd/2fda | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1zpbC  1zpcC  1zhrS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 26752.369 Da / Num. of mol.: 1 / Fragment: light chain, catalytic domain, residues 388-625 / Mutation: S75A, K78A, T115A, C123S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: F11 / Production host:  Pichia pastoris (fungus) / Strain (production host): X-33 / References: UniProt: P03951, coagulation factor XIa Pichia pastoris (fungus) / Strain (production host): X-33 / References: UniProt: P03951, coagulation factor XIa | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | 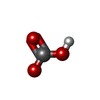  Mass: 61.017 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CHO3 / Comment: pH buffer*YM Mass: 61.017 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CHO3 / Comment: pH buffer*YM#3: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-682 / |   Mass: 385.485 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H27N7O3S Mass: 385.485 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H27N7O3S#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 255 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 255 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.78 Å3/Da / Density % sol: 55.76 % |
|---|---|
| Crystal grow | Temperature: 283 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: 2.0 M AMMONIUM SULFATE, 0.1 M TRIS-HCL, pH 8.5, VAPOR DIFFUSION, HANGING DROP, temperature 283K |
-Data collection
| Diffraction | Mean temperature: 123 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Jun 16, 2002 / Details: Blue Osmic Mirrors |
| Radiation | Monochromator: Blue Osmic Mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→28.6 Å / Num. all: 20224 / Num. obs: 20224 / % possible obs: 100 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 7.2 % / Biso Wilson estimate: 16.6 Å2 / Rmerge(I) obs: 0.063 / Net I/σ(I): 29.8 |
| Reflection shell | Resolution: 2→2.07 Å / Rmerge(I) obs: 0.335 / Mean I/σ(I) obs: 5.9 / Num. unique all: 1997 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB entry 1ZHR Resolution: 2→28.6 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 369696.48 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 51.3825 Å2 / ksol: 0.360468 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→28.6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.07 Å / Rfactor Rfree error: 0.017 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
