- PDB-2doh: The X-ray crystallographic structure of the angiogenesis inhibito... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 2doh
Title
The X-ray crystallographic structure of the angiogenesis inhibitor, angiostatin, bound a to a peptide from the group A streptococcal surface protein PAM
Components
Angiostatin
Plasminogen-binding group A streptococcal M-like protein PAM
Keywords
HYDROLASE / lysine-binding site / plasminogen / kringle domains
Function / homology
Function and homology information
plasmin / trans-synaptic signaling by BDNF, modulating synaptic transmission / trophoblast giant cell differentiation / tissue remodeling / tissue regeneration / Signaling by PDGF / positive regulation of fibrinolysis / mononuclear cell migration / negative regulation of cell-cell adhesion mediated by cadherin / protein antigen binding ...plasmin / trans-synaptic signaling by BDNF, modulating synaptic transmission / trophoblast giant cell differentiation / tissue remodeling / tissue regeneration / Signaling by PDGF / positive regulation of fibrinolysis / mononuclear cell migration / negative regulation of cell-cell adhesion mediated by cadherin / protein antigen binding / Dissolution of Fibrin Clot / myoblast differentiation / labyrinthine layer blood vessel development / plasminogen activation / biological process involved in interaction with symbiont / muscle cell cellular homeostasis / Activation of Matrix Metalloproteinases / extracellular matrix disassembly / negative regulation of fibrinolysis / negative regulation of cell-substrate adhesion / apolipoprotein binding / positive regulation of blood vessel endothelial cell migration / fibrinolysis / Degradation of the extracellular matrix / platelet alpha granule lumen / serine-type peptidase activity / protein processing / Schaffer collateral - CA1 synapse / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / kinase binding / blood coagulation / Platelet degranulation / protein-folding chaperone binding / extracellular matrix / protease binding / endopeptidase activity / blood microparticle / signaling receptor binding / serine-type endopeptidase activity / external side of plasma membrane / negative regulation of cell population proliferation / protein domain specific binding / glutamatergic synapse / enzyme binding / cell surface / proteolysis / : / extracellular exosome / extracellular region / plasma membrane Similarity search - Function
Plasminogen ligand, VEK-30 / Plasminogen (Pg) ligand in fibrinolytic pathway / : / Streptococcal M proteins D repeats region profile. / : / Streptococcal M proteins C repeat profile. / Peptidase S1A, plasmin / Plasminogen Kringle 4 / Plasminogen Kringle 4 / divergent subfamily of APPLE domains ...Plasminogen ligand, VEK-30 / Plasminogen (Pg) ligand in fibrinolytic pathway / : / Streptococcal M proteins D repeats region profile. / : / Streptococcal M proteins C repeat profile. / Peptidase S1A, plasmin / Plasminogen Kringle 4 / Plasminogen Kringle 4 / divergent subfamily of APPLE domains / : / PAN/Apple domain profile. / PAN domain / PAN/Apple domain / YSIRK Gram-positive signal peptide / Kringle domain / Kringle / Kringle, conserved site / Kringle superfamily / Kringle domain signature. / Kringle domain profile. / Kringle domain / Kringle-like fold / Serine proteases, trypsin family, histidine active site / Serine proteases, trypsin family, serine active site / Serine proteases, trypsin family, histidine active site. / Peptidase S1A, chymotrypsin family / Serine proteases, trypsin family, serine active site. / Serine proteases, trypsin domain profile. / Trypsin-like serine protease / Serine proteases, trypsin domain / Trypsin / Peptidase S1, PA clan, chymotrypsin-like fold / Peptidase S1, PA clan / Beta Barrel / Mainly Beta Similarity search - Domain/homology
1,4-DIETHYLENE DIOXIDE / Plasminogen / Plasminogen-binding group A streptococcal M-like protein PAM Similarity search - Component
Type: MAR CCD 165 mm / Detector: CCD / Date: Mar 16, 2000
Radiation
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1 Å / Relative weight: 1
Reflection
Av σ(I) over netI: 12.4 / Number: 320127 / Rmerge(I) obs: 0.091 / Χ2: 2.53 / D res high: 2 Å / D res low: 50 Å / Num. obs: 22779 / % possible obs: 80.2
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads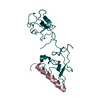








 PDBj
PDBj




 Assembly
Assembly
 Pichia pastoris (fungus) / References: UniProt: P00747, plasmin
Pichia pastoris (fungus) / References: UniProt: P00747, plasmin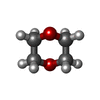
 Mass: 88.105 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H8O2
Mass: 88.105 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 18.015 Da / Num. of mol.: 311 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 311 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 17-ID / Wavelength: 1 Å
/ Beamline: 17-ID / Wavelength: 1 Å Processing
Processing