| Entry | Database: PDB / ID: 2c3b
|
|---|
| Title | The Crystal Structure of Aspergillus fumigatus Cyclophilin reveals 3D Domain Swapping of a Central Element |
|---|
 Components Components | PPIASE |
|---|
 Keywords Keywords | ISOMERASE / 3D DOMAIN SWAPPING / MISFOLDING / PPIASE / ASP F 11 / ALLERGEN / ROTAMASE |
|---|
| Function / homology |  Function and homology information Function and homology information
cyclosporin A binding / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / protein folding / identical protein binding / cytoplasmSimilarity search - Function Cyclophilin-like / Cyclophilin / Cyclophilin-type peptidyl-prolyl cis-trans isomerase / Cyclophilin-type peptidyl-prolyl cis-trans isomerase, conserved site / Cyclophilin-type peptidyl-prolyl cis-trans isomerase signature. / Cyclophilin-type peptidyl-prolyl cis-trans isomerase domain profile. / Cyclophilin-type peptidyl-prolyl cis-trans isomerase domain / Cyclophilin type peptidyl-prolyl cis-trans isomerase/CLD / Cyclophilin-like domain superfamily / Beta Barrel / Mainly BetaSimilarity search - Domain/homology |
|---|
| Biological species |   ASPERGILLUS FUMIGATUS (mold) ASPERGILLUS FUMIGATUS (mold) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.85 Å |
|---|
 Authors Authors | Limacher, A. / Kloer, D.P. / Fluckiger, S. / Folkers, G. / Crameri, R. / Scapozza, L. |
|---|
 Citation Citation | Journal: Structure / Year: 2006
Title: The Crystal Structure of Aspergillus Fumigatus Cyclophilin Reveals 3D Domain Swapping of a Central Element
Authors: Limacher, A. / Kloer, D.P. / Fluckiger, S. / Folkers, G. / Crameri, R. / Scapozza, L. |
|---|
| History | | Deposition | Oct 5, 2005 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Jan 30, 2006 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jul 13, 2011 | Group: Advisory / Version format compliance |
|---|
| Revision 1.2 | Nov 13, 2024 | Group: Data collection / Database references ...Data collection / Database references / Other / Structure summary
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_entry_details / pdbx_modification_feature
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf |
|---|
|
|---|
| Remark 650 | HELIX DETERMINATION METHOD: AUTHOR PROVIDED. |
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj

 Assembly
Assembly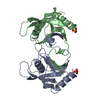


 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 123 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 123 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X06SA / Wavelength: 0.9999
/ Beamline: X06SA / Wavelength: 0.9999  Processing
Processing