 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2bq1: Ribonucleotide reductase class 1b holocomplex R1E,R2F from Salmon... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2bq1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Ribonucleotide reductase class 1b holocomplex R1E,R2F from Salmonella typhimurium | ||||||
 Components Components |
| ||||||
 Keywords Keywords | OXIDOREDUCTASE / R1 / R2 / R1E / R2F / IRON / CLASS 1B / HOLOCOMPLEX / ALLOSTERIC REGULATION / RIBONUCLEOTIDE REDUCTASE / ATP-BINDING / METAL-BINDING / DNA REPLICATION / RADICAL TRANSFER / ALLOSTERIC ENZYME / ASYMMETRIC COMPLEX / NUCLEOTIDE-BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationribonucleoside-diphosphate reductase complex / ribonucleoside-diphosphate reductase / ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor / deoxyribonucleotide biosynthetic process / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  SALMONELLA TYPHIMURIUM (bacteria) SALMONELLA TYPHIMURIUM (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.99 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.99 Å | ||||||
 Authors Authors | Uppsten, M. / Farnegardh, M. / Domkin, V. / Uhlin, U. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2006 Title: The First Holocomplex Structure of Ribonucleotide Reductase Gives New Insight Into its Mechanism of Action Authors: Uppsten, M. / Farnegardh, M. / Domkin, V. / Uhlin, U. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2bq1.cif.gz | 386.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2bq1.ent.gz | 312.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2bq1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 2bq1_validation.pdf.gz | 1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 2bq1_full_validation.pdf.gz | 1.1 MB | Display | |
| Data in XML | 2bq1_validation.xml.gz | 69.9 KB | Display | |
| Data in CIF | 2bq1_validation.cif.gz | 93.8 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bq/2bq1ftp://data.pdbj.org/pub/pdb/validation_reports/bq/2bq1 | HTTPS FTP |
-Related structure data
| Related structure data |  1pemS S: Starting model for refinement |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 80686.211 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) SALMONELLA TYPHIMURIUM (bacteria) / Plasmid: PET24A / Production host: References: UniProt: Q08698, ribonucleoside-diphosphate reductase #2: Protein | Mass: 36262.988 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) SALMONELLA TYPHIMURIUM (bacteria) / Production host: References: UniProt: P17424, ribonucleoside-diphosphate reductase #3: Chemical | 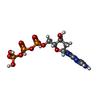  Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3#4: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#5: Chemical | ChemComp-FE /   Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: FeCompound details | CATALYTIC ACTIVITY: 2'-DEOXYRIBONUCLEOSIDE DIPHOSPHATE + THIOREDOXIN DISULFIDE + H(2)O = ...CATALYTIC ACTIVITY: 2'-DEOXYRIBON | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.79 Å3/Da / Density % sol: 66 % |
|---|---|
| Crystal grow | pH: 7.5 Details: 1:1 MIX OF PROTEIN SOLUTION (8 MG/ML R1E, 4 MG/ML R2F, 1.5 MM DGTP, 1.5 MM ADP, 1.5 MM MGCL2 AND 1.5 MM HYDROXYUREA) AND RESERVOIR SOLUTION CONSISTING OF 100 MM NAHEPES PH 7.5 AND 1.8 M SODIUM FORMATE. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.934 / Beamline: ID14-1 / Wavelength: 0.934 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Apr 23, 2004 |
| Radiation | Monochromator: DIAMOND (111), GE(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.934 Å / Relative weight: 1 |
| Reflection | Resolution: 4→40 Å / Num. obs: 29196 / % possible obs: 99.8 % / Observed criterion σ(I): 1 / Redundancy: 15.1 % / Rmerge(I) obs: 0.15 / Net I/σ(I): 13.5 |
| Reflection shell | Resolution: 4→4.07 Å / Redundancy: 7.2 % / Rmerge(I) obs: 0.39 / Mean I/σ(I) obs: 4 / % possible all: 98.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1PEM Resolution: 3.99→182.57 Å / Cor.coef. Fo:Fc: 0.811 / Cor.coef. Fo:Fc free: 0.778 / SU B: 53.692 / SU ML: 0.757 / Cross valid method: THROUGHOUT / ESU R Free: 0.946 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 65.31 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.99→182.57 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|