 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2act: CRYSTALLOGRAPHIC REFINEMENT OF THE STRUCTURE OF ACTINIDIN AT 1.7 ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2act | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTALLOGRAPHIC REFINEMENT OF THE STRUCTURE OF ACTINIDIN AT 1.7 ANGSTROMS RESOLUTION BY FAST FOURIER LEAST-SQUARES METHODS | |||||||||
 Components Components | ACTINIDIN PRECURSOR | |||||||||
 Keywords Keywords | HYDROLASE (PROTEINASE) | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  Actinidia chinensis (golden kiwifruit) Actinidia chinensis (golden kiwifruit) | |||||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.7 Å X-RAY DIFFRACTION / Resolution: 1.7 Å | |||||||||
 Authors Authors | Baker, E.N. | |||||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.A / Year: 1980 Title: Crystallographic Refinement of the Structure of Actinidin at 1.7 Angstroms Resolution by Fast Fourier Least-Squares Methods Authors: Baker, E.N. / Dodson, E.J. #1: Journal: J.Mol.Biol. / Year: 1980Title: Structure of Actinidin, After Refinement at 1.7 Angstroms Resolution Authors: Baker, E.N. #2: Journal: J.Mol.Biol. / Year: 1977Title: Structure of Actinidin. Details of the Polypeptide Chain Conformation and Active Site from an Electron Density Map at 2.8 Angstroms Resolution Authors: Baker, E.N. #3: Journal: Biochem.J. / Year: 1978Title: The Amino Acid Sequence of the Tryptic Peptides from Actinidin, a Proteolytic Enzyme from the Fruit of Actinidia Chinensis Authors: Carne, A. / Moore, C.H. | |||||||||
| History |
| |||||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE ...SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. STRANDS 2, 3, 4, 5 OF B1 AND B2 ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2act.cif.gz | 61 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2act.ent.gz | 43.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2act.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ac/2actftp://data.pdbj.org/pub/pdb/validation_reports/ac/2act | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUES 9, 42, 44, 58 AND 129 ARE DISORDERED. / 2: SEE REMARK 10. 3: AMINO ACID SEQUENCE ANALYSIS IDENTIFIES RESIDUE 86 AS ASP, BUT REFINEMENT SHOWS IT TO BE GLN OR GLU (HERE TAKEN AS GLU). 4: NO DENSITY BEYOND CB. / 5: POORLY DEFINED BEYOND CB. 6: ELECTRON DENSITY PEAK 2 ANGSTROMS FROM CB. COULD BE SER (I.E. SEQUENCE ERROR). PEAK CURRENTLY IDENTIFIED AS HOH 92. 7: POORLY DEFINED SIDECHAIN. / 8: CIS-PROLINE. 9: REFINED POORLY. COULD BE SEQUENCE ERROR. SHOULD PERHAPS BE VAL. 10: VERY WEAK DENSITY. |
-Components
| #1: Protein | Mass: 23718.986 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Actinidia chinensis (golden kiwifruit) / References: UniProt: P00785 |
|---|---|
| #2: Chemical | ChemComp-NH4 / 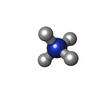  Mass: 18.038 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H4N Mass: 18.038 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H4N |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 272 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 272 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Nonpolymer details | ALL SOLVENT MOLECULES ARE REGARDED AS WATER, WITH THE EXCEPTION OF NH4 4 WHICH IS BELIEVED TO BE AN ...ALL SOLVENT MOLECULES ARE REGARDED AS WATER, WITH THE EXCEPTION OF NH4 4 WHICH IS BELIEVED TO BE AN AMMONIUM ION AND HAS THREE HYDROGEN-BOND ACCEPTORS AS NEAREST NEIGHBORS. SOLVENT MOLECULES ARE NUMBERED IN ORDER OF THEIR RELIABILIT |
| Sequence details | AMINO ACID SEQUENCE ANALYSIS IDENTIFIES RESIDUE 86 AS ASP, BUT REFINEMENT SHOWS IT TO BE GLN OR GLU ...AMINO ACID SEQUENCE ANALYSIS IDENTIFIES |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.83 % |
|---|---|
| Crystal grow | *PLUS Method: other / Details: Baker, E.N., (1974) J. Mol. Bol., 74, 411. |
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.7 Å / Num. obs: 24157 / Num. measured all: 47000 / Rmerge(I) obs: 0.056 |
- Processing
Processing
| Software | Name: FAST-FOURIER / Version: LEAST-SQUARES REFINEMENT / Classification: refinement | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.7→10 Å Details: ACCURACY OF ATOMIC COORDINATES (DERIVED FROM INVERSE LEAST-SQUARES MATRIX) IS GIVEN BELOW AS A FUNCTION OF ATOMIC B VALUE. B(ANGSTROMS SQUARED) SIGMA 0 - 5 0.04 ANGSTROMS 5 - 10 0.05 ...Details: ACCURACY OF ATOMIC COORDINATES (DERIVED FROM INVERSE LEAST-SQUARES MATRIX) IS GIVEN BELOW AS A FUNCTION OF ATOMIC B VALUE. B(ANGSTROMS SQUARED) SIGMA 0 - 5 0.04 ANGSTROMS 5 - 10 0.05 ANGSTROMS 10 - 15 0.06 ANGSTROMS 15 - 20 0.07 ANGSTROMS OVER 20 0.08 ANGSTROMS OVERALL 0.06 ANGSTROMS ATOMS IN ASN AND GLN SIDE CHAINS ARE LABELLED AS OD1 AND ND2 OR OE1 AND NE2 RESPECTIVELY, THE CHOICE BEING BASED ON ENVIRONMENT (E.G. H-BONDS) AND/OR B VALUES.
| ||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→10 Å
| ||||||||||||
| Refinement | *PLUS Num. reflection all: 23990 / Num. reflection obs: 19724 / σ(I): 2 / Rfactor all: 0.171 / Rfactor obs: 0.152 | ||||||||||||
| Solvent computation | *PLUS | ||||||||||||
| Displacement parameters | *PLUS |
