- PDB-1yb3: Conserved hypothetical protein Pfu-178653-001 from Pyrococcus furiosus -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 1yb3
Title
Conserved hypothetical protein Pfu-178653-001 from Pyrococcus furiosus
Components
hypothetical protein
Keywords
STRUCTURAL GENOMICS / UNKNOWN FUNCTION / Protein Structure Initiative / PSI / conserved hypothetical protein / Pyrococcus furiosus / hyperthermophile / SECSG / The Southeast Collaboratory for Structural Genomics
Function / homology
Protein of unknown function DUF3201 / Protein of unknown function (DUF3201) / Bira Bifunctional Protein; Domain 2 / BirA Bifunctional Protein; domain 2 / Class II Aminoacyl-tRNA synthetase/Biotinyl protein ligase (BPL) and lipoyl protein ligase (LPL) / 2-Layer Sandwich / Alpha Beta / DUF3201 domain-containing protein
Function and homology information
Biological species
Pyrococcus furiosus (archaea)
Method
X-RAY DIFFRACTION / SYNCHROTRON / SAS / sad / Resolution: 1.6 Å
BIOMOLECULE THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 1 CHAIN. THE ...BIOMOLECULE THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 1 CHAIN. THE BIOLOGICAL UNIT IS UNKNOWN.
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.9795 Å / Relative weight: 1
Reflection
Resolution: 1.6→50 Å / Num. obs: 26254 / % possible obs: 84.8 % / Rmerge(I) obs: 0.052 / Χ2: 1.337
Reflection shell
Resolution (Å)
Rmerge(I) obs
Num. measured all
Χ2
Diffraction-ID
% possible all
1.6-1.66
0.218
1066
1.013
1
34.6
1.66-1.72
0.2
1595
1.132
1
51.6
1.72-1.8
0.195
2356
0.915
1
77.3
1.8-1.9
0.165
2925
1.026
1
95
1.9-2.02
0.128
3022
1.157
1
97.6
2.02-2.17
0.085
3007
1.308
1
97.9
2.17-2.39
0.067
3031
1.5
1
98
2.39-2.74
0.058
3049
1.54
1
98.3
2.74-3.45
0.049
3074
1.822
1
98.4
3.45-50
0.042
3129
1.231
1
98.2
-
Phasing
Phasing
Method: sad
Phasing MAD
D res high: 2.4 Å / D res low: 20 Å / FOM : 0.39 / Reflection: 9009
Phasing MAD shell
Resolution (Å)
FOM
Reflection
8.22-20
0.42
470
5.33-8.22
0.44
764
4.21-5.33
0.4
968
3.59-4.21
0.44
1125
3.18-3.59
0.45
1254
2.88-3.18
0.41
1367
2.66-2.88
0.38
1487
2.48-2.66
0.28
1574
-
Processing
Software
Name
Version
Classification
NB
DENZO
datareduction
SCALEPACK
datascaling
SOLVE
2.06
phasing
RESOLVE
2.06
phasing
REFMAC
refmac_5.2.0005
refinement
PDB_EXTRACT
1
dataextraction
MAR345
datacollection
ISAS
phasing
ARP/wARP
modelbuilding
Refinement
Method to determine structure: SAS / Resolution: 1.6→48.45 Å / Cor.coef. Fo:Fc: 0.957 / Cor.coef. Fo:Fc free: 0.943 / WRfactor Rfree: 0.245 / WRfactor Rwork: 0.217 / SU B: 1.663 / SU ML: 0.06 / SU R Cruickshank DPI: 0.1 / Cross valid method: THROUGHOUT / ESU R: 0.1 / ESU R Free: 0.097 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2291
1312
4.997 %
RANDOM
Rwork
0.2039
-
-
-
all
0.205
-
-
-
obs
0.20521
26254
84.792 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT
Displacement parameters
Biso mean: 26.965 Å2
Baniso -1
Baniso -2
Baniso -3
1-
-1.491 Å2
0 Å2
-0.15 Å2
2-
-
0.988 Å2
0 Å2
3-
-
-
0.359 Å2
Refinement step
Cycle: LAST / Resolution: 1.6→48.45 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1392
0
1
102
1495
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.012
0.022
1461
X-RAY DIFFRACTION
r_angle_refined_deg
1.151
1.954
1974
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
5.31
5
165
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
27.983
25
88
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
12.034
15
258
X-RAY DIFFRACTION
r_dihedral_angle_4_deg
17.644
15
7
X-RAY DIFFRACTION
r_chiral_restr
0.087
0.2
202
X-RAY DIFFRACTION
r_gen_planes_refined
0.005
0.02
1150
X-RAY DIFFRACTION
r_nbd_refined
0.194
0.2
642
X-RAY DIFFRACTION
r_nbtor_refined
0.309
0.2
1009
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.098
0.2
76
X-RAY DIFFRACTION
r_symmetry_vdw_refined
0.212
0.2
44
X-RAY DIFFRACTION
r_symmetry_hbond_refined
0.118
0.2
7
X-RAY DIFFRACTION
r_mcbond_it
2.373
2
873
X-RAY DIFFRACTION
r_mcangle_it
3.04
3
1352
X-RAY DIFFRACTION
r_scbond_it
2.437
2
690
X-RAY DIFFRACTION
r_scangle_it
3.637
3
622
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Num. reflection all
% reflection obs (%)
1.6-1.639
0.324
39
0.282
718
2276
33.26
1.639-1.684
0.326
44
0.283
910
2209
43.187
1.684-1.733
0.308
59
0.286
1223
2167
59.16
1.733-1.786
0.337
78
0.272
1536
2089
77.262
1.786-1.845
0.29
89
0.257
1774
2027
91.909
1.845-1.909
0.284
86
0.222
1811
1957
96.934
1.909-1.981
0.231
95
0.216
1746
1888
97.511
1.981-2.062
0.27
86
0.201
1703
1832
97.653
2.062-2.154
0.218
92
0.196
1632
1761
97.899
2.154-2.259
0.245
91
0.196
1555
1677
98.151
2.259-2.38
0.237
72
0.202
1498
1601
98.064
2.38-2.524
0.236
69
0.212
1407
1503
98.204
2.524-2.698
0.264
67
0.223
1335
1427
98.248
2.698-2.914
0.227
72
0.22
1236
1331
98.272
2.914-3.191
0.216
67
0.205
1150
1234
98.622
3.191-3.565
0.205
48
0.191
1038
1101
98.638
3.565-4.113
0.185
54
0.181
921
992
98.286
4.113-5.028
0.174
48
0.156
786
848
98.349
5.028-7.073
0.263
34
0.207
620
656
99.695
7.073-48.45
0.24
22
0.217
343
387
94.315
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Pyrococcus furiosus (archaea)
Pyrococcus furiosus (archaea) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads





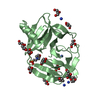



 PDBj
PDBj
 Assembly
Assembly



 Num. of mol.: 1 / Source method: obtained synthetically
Num. of mol.: 1 / Source method: obtained synthetically Mass: 18.015 Da / Num. of mol.: 102 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 102 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 22-ID / Wavelength: 0.9795 Å
/ Beamline: 22-ID / Wavelength: 0.9795 Å Processing
Processing