 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1wat: THE THREE-DIMENSIONAL STRUCTURE OF THE LIGAND-BINDING DOMAIN OF A... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1wat | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE THREE-DIMENSIONAL STRUCTURE OF THE LIGAND-BINDING DOMAIN OF A WILD-TYPE BACTERIAL CHEMOTAXIS RECEPTOR | ||||||
 Components Components | ASPARTATE RECEPTOR | ||||||
 Keywords Keywords | CHEMOTAXIS | ||||||
| Function / homology |  Function and homology information Function and homology informationchemotaxis / transmembrane signaling receptor activity / signal transduction / plasma membrane Similarity search - Function | ||||||
| Biological species |  Salmonella typhimurium (bacteria) Salmonella typhimurium (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 3 Å X-RAY DIFFRACTION / Resolution: 3 Å | ||||||
 Authors Authors | Kim, S.-H. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1993 Title: The three-dimensional structure of the ligand-binding domain of a wild-type bacterial chemotaxis receptor. Structural comparison to the cross-linked mutant forms and conformational changes upon ligand binding. Authors: Yeh, J.I. / Biemann, H.P. / Pandit, J. / Koshland, D.E. / Kim, S.H. #1: Journal: Science / Year: 1991Title: Three-Dimensional Structures of the Ligand-Binding Domain of the Bacterial Aspartate Receptor with and without a Ligand Authors: Milburn, M.V. / Prive, G.G. / Milligan, D.L. / Scott, W.G. / Yeh, J.I. / Jancarik, J. / Koshland Junior, D.E. / Kim, S.-H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1wat.cif.gz | 58.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1wat.ent.gz | 41.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1wat.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wa/1watftp://data.pdbj.org/pub/pdb/validation_reports/wa/1wat | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: THR A 111 - PRO A 112 OMEGA = 111.59 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 2: LEU A 113 - PRO A 114 OMEGA = 211.53 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 3: THR B 179 - PHE B 180 OMEGA = 84.24 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 4: THE LOOP REGIONS, RESIDUES A 76 - A 84 AND B 76 - B 84, ARE DISORDERED. | ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.8583, 0.439267, 0.2652), Vector: Details | THE TRANSFORMATION PRESENTED ON *MTRIX* RECORDS BELOW WILL GENERATE APPROXIMATE COORDINATES FOR CHAIN *A* WHEN APPLIED TO CHAIN *B*. THIS CORRESPONDS TO A ROTATION OF 178.2 DEGREES, WHICH IS AN APPROXIMATE (MOLECULAR) TWO-FOLD. | |
-Components
| #1: Protein | Mass: 16366.229 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Salmonella typhimurium (bacteria) / References: UniProt: P02941#2: Chemical | ChemComp-ASP / | 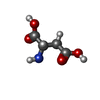  Type: L-peptide linking / Mass: 133.103 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H7NO4 Type: L-peptide linking / Mass: 133.103 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H7NO4 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.34 Å3/Da / Density % sol: 71.68 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Reflection | *PLUS Highest resolution: 3 Å / Num. obs: 7114 / % possible obs: 62 % / Num. measured all: 15061 / Rmerge(I) obs: 0.071 |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Highest resolution: 3 Å / σ(F): 1.5 Details: THE LOOP REGIONS, RESIDUES A 76 - A 84 AND B 76 - B 84, ARE DISORDERED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 3 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.192 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 32.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d / Dev ideal: 3.696 |
