 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
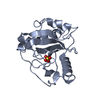
Yorodumi- PDB-1vmg: Crystal structure of MazG nucleotide pyrophosphohydrolase (138166... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1vmg | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of MazG nucleotide pyrophosphohydrolase (13816655) from Sulfolobus solfataricus at 1.46 A resolution | ||||||
 Components Components | Hypothetical protein SSO3215 | ||||||
 Keywords Keywords | HYDROLASE / 13816655 / MazG nucleotide pyrophosphohydrolase / Structural Genomics / JCSG / Protein Structure Initiative / PSI / Joint Center for Structural Genomics | ||||||
| Function / homology | NTP pyrophosphohydrolase MazG, putative catalytic core / MazG nucleotide pyrophosphohydrolase domain / MazG-like / Helix Hairpins / Orthogonal Bundle / Mainly Alpha / : / Unknown ligand / NTP pyrophosphohydrolase MazG-like domain-containing protein Function and homology information Function and homology information | ||||||
| Biological species |   Sulfolobus solfataricus (archaea) Sulfolobus solfataricus (archaea) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.46 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.46 Å | ||||||
 Authors Authors | Joint Center for Structural Genomics (JCSG) | ||||||
 Citation Citation | Journal: To be published Title: Crystal structure of MazG nucleotide pyrophosphohydrolase (13816655) from Sulfolobus solfataricus at 1.46 A resolution Authors: Joint Center for Structural Genomics (JCSG) | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1vmg.cif.gz | 34.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1vmg.ent.gz | 23.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1vmg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vm/1vmgftp://data.pdbj.org/pub/pdb/validation_reports/vm/1vmg | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |









-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 11182.214 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Sulfolobus solfataricus (archaea) / Strain: P2 / Production host:  References: UniProt: Q97U11, Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides |
|---|---|
| #2: Chemical | ChemComp-LI /   Mass: 6.941 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Li Mass: 6.941 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Li |
| #3: Chemical | ChemComp-UNL / Num. of mol.: 1 / Source method: obtained synthetically |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 106 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 106 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.3 Å3/Da / Density % sol: 62.4 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop, nanodrop / pH: 7.5 Details: 1.5M Li2SO4, 0.1M HEPES pH 7.5, VAPOR DIFFUSION,SITTING DROP,NANODROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.3.1 / Wavelength: 0.979834 / Wavelength: 0.979834,0.979694,1.020035 / Beamline: 8.3.1 / Wavelength: 0.979834 / Wavelength: 0.979834,0.979694,1.020035 | ||||||||||||
| Detector | Type: ADSC / Detector: CCD / Date: Sep 18, 2004 | ||||||||||||
| Radiation | Monochromator: Double Crystal Si(111) / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 1.46→25.62 Å / Num. obs: 25360 / % possible obs: 93.3 % / Redundancy: 5.9 % / Biso Wilson estimate: 23.08 Å2 / Rsym value: 0.044 / Net I/σ(I): 19.6 | ||||||||||||
| Reflection shell | Resolution: 1.46→1.54 Å / Redundancy: 2 % / Mean I/σ(I) obs: 2.2 / Num. unique all: 2241 / Rsym value: 0.435 / % possible all: 57.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.46→25.62 Å / Cor.coef. Fo:Fc: 0.976 / Cor.coef. Fo:Fc free: 0.975 / SU B: 1.453 / SU ML: 0.027 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.043 / ESU R Free: 0.041 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS 2. AN UNKNOWN ENTITY BETWEEN TYR 16 AND TRP 61 WAS AS UNK, UNKNOWN LIGAND. THE DENSITY LOOKS SIMILAR TO A GUANINE BASE. 3. THERE ARE ...Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS 2. AN UNKNOWN ENTITY BETWEEN TYR 16 AND TRP 61 WAS AS UNK, UNKNOWN LIGAND. THE DENSITY LOOKS SIMILAR TO A GUANINE BASE. 3. THERE ARE ADDITIONAL UNEXPLAINED DENSITIES NEAR ARG 23, TRP 31, TRP 61. IT MAY BE RELATED TO UNK, HOWEVER, DENSITY IS TOO FRAGMENTED TO IDENTIFY OR MODEL. 4. A LI ION WAS TENATIVELY MODELLED ACCORDING TO THE ENVIROMENT. AND ITS PRESENCE IN THE CRYSTALLIZATION BUFFER.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.062 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.46→25.62 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.46→1.498 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 68.539 Å / Origin y: 7.845 Å / Origin z: 3.915 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection: ALL |
