 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ufy | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal analysis of chorismate mutase from thermus thermophilus | ||||||
 Components Components | Chorismate mutase | ||||||
 Keywords Keywords | ISOMERASE / CHORISMATE MUTASE / SHIKIMATE PATHWAY / MUTANT / RIKEN Structural Genomics/Proteomics Initiative / RSGI / Structural Genomics | ||||||
| Function / homology |  Function and homology information Function and homology informationchorismate metabolic process / chorismate mutase / chorismate mutase activity / aromatic amino acid biosynthetic process / amino acid biosynthetic process / cytoplasm Similarity search - Function | ||||||
| Biological species |   Thermus thermophilus (bacteria) Thermus thermophilus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.96 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.96 Å | ||||||
 Authors Authors | Inagaki, E. / Miyano, M. / Tahirov, T.H. / RIKEN Structural Genomics/Proteomics Initiative (RSGI) | ||||||
 Citation Citation | Journal: To be Published Title: The Crystal Structure of Chorismate Mutase from Thermus Thermophilus Authors: Inagaki, E. / Kuramitsu, S. / Yokoyama, S. / Miyano, M. / Tahirov, T.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ufy.cif.gz | 69.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ufy.ent.gz | 51.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1ufy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/uf/1ufyftp://data.pdbj.org/pub/pdb/validation_reports/uf/1ufy | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1odeSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||||||||
| Details | The biological assembly is a trimer generated from the monomer in the asymmetric unit by the operations: -y+2, x-y+1, z and -x+y+1, -x+1, z. |
-Components
| #1: Protein | Mass: 13666.668 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermus thermophilus (bacteria) / Strain: HB8 / Plasmid: PET11A / Species (production host): Escherichia coli / Production host: | ||
|---|---|---|---|
| #2: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl | ||
| #3: Chemical | ChemComp-MLI / 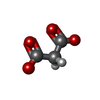  Mass: 102.046 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2O4 Mass: 102.046 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2O4 | ||
| #4: Chemical | ChemComp-MES /   Mass: 195.237 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 195.237 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 132 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 132 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.71 Å3/Da / Density % sol: 54.7 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: microbatch / pH: 7 Details: SODIUM MALONATE, MES, pH 7.0, MICRO BATCH, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL26B1 / Wavelength: 0.8 / Wavelength: 0.8 Å / Beamline: BL26B1 / Wavelength: 0.8 / Wavelength: 0.8 Å |
| Detector | Type: RIGAKU RAXIS V / Detector: IMAGE PLATE / Date: Nov 7, 2002 / Details: mirrors |
| Radiation | Monochromator: Si / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8 Å / Relative weight: 1 |
| Reflection | Resolution: 0.95→50 Å / Num. all: 93458 / Num. obs: 90841 / % possible obs: 97.2 % / Observed criterion σ(I): -1 / Redundancy: 4.7 % / Biso Wilson estimate: 2.7 Å2 / Rmerge(I) obs: 0.05 / Net I/σ(I): 22.2 |
| Reflection shell | Resolution: 0.95→0.96 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.345 / Mean I/σ(I) obs: 1.5 / % possible all: 59.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PROTOMER A OF PDB ENTRY 1ODE Resolution: 0.96→10 Å / Num. parameters: 10275 / Num. restraintsaints: 12599 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH & HUBER Details: Anisotropic refinement reduced free R (no cutoff) by 0.03. In final 20 cycles of refinement all refrections were included. Free R value is caluculated before including all refrections in the refinement.
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-228 | |||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.11 Å / Luzzati d res low obs: 3.84 Å / Num. disordered residues: 5 / Occupancy sum hydrogen: 963.52 / Occupancy sum non hydrogen: 1043.55 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.96→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 0.96→1 Å /
|
