 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1r4a: Crystal Structure of GTP-bound ADP-ribosylation Factor Like Prote... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1r4a | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of GTP-bound ADP-ribosylation Factor Like Protein 1 (Arl1) and GRIP Domain of Golgin245 COMPLEX | ||||||
 Components Components |
| ||||||
 Keywords Keywords | PROTEIN TRANSPORT / Ras-like G Protein structure / Three-helix GRIP domain | ||||||
| Function / homology |  Function and homology information Function and homology informationphospholipase D activator activity / Retrograde transport at the Trans-Golgi-Network / Retrograde transport at the Trans-Golgi-Network / toxin metabolic process / Golgi vesicle transport / Signaling by membrane-tethered fusions of PDGFRA or PDGFRB / protein localization to Golgi apparatus / Golgi to plasma membrane protein transport / retrograde transport, endosome to Golgi / Golgi organization ...phospholipase D activator activity / Retrograde transport at the Trans-Golgi-Network / Retrograde transport at the Trans-Golgi-Network / toxin metabolic process / Golgi vesicle transport / Signaling by membrane-tethered fusions of PDGFRA or PDGFRB / protein localization to Golgi apparatus / Golgi to plasma membrane protein transport / retrograde transport, endosome to Golgi / Golgi organization / positive regulation of axon extension / vesicle-mediated transport / trans-Golgi network / intracellular protein transport / small GTPase binding / enzyme activator activity / GTPase binding / Golgi membrane / protein domain specific binding / GTPase activity / GTP binding / Golgi apparatus / extracellular exosome / nucleoplasm / metal ion binding / plasma membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |   Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Wu, M. / Lu, L. / Hong, W. / Song, H. | ||||||
 Citation Citation | Journal: Nat.Struct.Mol.Biol. / Year: 2004 Title: Structural basis for recruitment of GRIP domain golgin-245 by small GTPase Arl1. Authors: Wu, M. / Lu, L. / Hong, W. / Song, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1r4a.cif.gz | 191 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1r4a.ent.gz | 151.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1r4a.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/r4/1r4aftp://data.pdbj.org/pub/pdb/validation_reports/r4/1r4a | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ksgS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | Four molecules are generated by two 2-fold symmetry operations. / Tetramer is generated by two 2-fold symmetry operations. |
-Components
| #1: Protein | Mass: 18737.615 Da / Num. of mol.: 4 / Fragment: Arl1 (Residue 16-180) Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Protein | Mass: 6198.274 Da / Num. of mol.: 4 / Fragment: GRIP Domain (Residue 2172-2222) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GOLGIN-245 / Plasmid: pGEX-6p-1 / Production host: #3: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#4: Chemical | ChemComp-GNP / 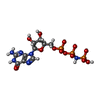  Mass: 522.196 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H17N6O13P3 Mass: 522.196 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H17N6O13P3Comment: GppNHp, GMPPNP, energy-carrying molecule analogue*YM #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 268 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 268 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 52.35 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8 Details: PEG3350, potassium thiocyanate, pH 8.0, VAPOR DIFFUSION, HANGING DROP, temperature 293.0K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7A / Wavelength: 0.972 Å / Beamline: BW7A / Wavelength: 0.972 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jul 10, 2003 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.972 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→99.9 Å / Num. all: 55269 / Num. obs: 55268 / % possible obs: 91 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.1 % / Rmerge(I) obs: 0.075 / Rsym value: 0.075 / Net I/σ(I): 8.4 |
| Reflection shell | Resolution: 2.1→2.18 Å / Redundancy: 2.3 % / Rmerge(I) obs: 0.539 / Mean I/σ(I) obs: 1.88 / Rsym value: 0.479 / % possible all: 56.5 |
| Reflection | *PLUS Lowest resolution: 39 Å / % possible obs: 97.1 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1KSG Resolution: 2.3→20 Å / Cross valid method: free R / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber / Details: The structure was refined also with CNS(BRUNGER)
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→20 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.39 Å / Total num. of bins used: 20 /
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Version: 5 / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.26 / Rfactor Rwork: 0.245 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
