 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qd9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Bacillus subtilis YABJ | ||||||
 Components Components | PURINE REGULATORY PROTEIN YABJ | ||||||
 Keywords Keywords | GENE REGULATION / PERCHLORIC ACID SOLUBLE PROTEIN / PURINE REGULATION / YJGF/YER057C FAMILY | ||||||
| Function / homology |  Function and homology information Function and homology information2-oxobutyrate biosynthetic process / 2-iminobutanoate/2-iminopropanoate deaminase / 2-iminobutanoate deaminase activity / 2-iminopropanoate deaminase activity / deaminase activity / : / response to toxic substance / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / HG MAD / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / HG MAD / Resolution: 1.7 Å | ||||||
 Authors Authors | Smith, J.L. / Sinha, S. / Rappu, P. / Lange, S.C. / Mantsala, P. / Zalkin, H. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1999 Title: Crystal structure of Bacillus subtilis YabJ, a purine regulatory protein and member of the highly conserved YjgF family. Authors: Sinha, S. / Rappu, P. / Lange, S.C. / Mantsala, P. / Zalkin, H. / Smith, J.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qd9.cif.gz | 94.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qd9.ent.gz | 72.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1qd9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qd/1qd9ftp://data.pdbj.org/pub/pdb/validation_reports/qd/1qd9 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj


















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.0631, -0.4353, 0.8981), Vector: Details | THE MODEL INCLUDES RESIDUES 2 - 125 IN EACH SUBUNIT. N-TERMINAL SEQUENCE ANALYSIS INDICATED THAT MET1 WAS NOT PRESENT IN THE PURIFIED PROTEIN USED FOR CRYSTALLIZATION. YABJ IS A TRIMER. SUBUNITS IN THE TRIMER ARE RELATED BY NCS THREE-FOLD ROTATIONAL SYMMETRY. THE ASYMMETRIC UNIT CONTAINS ONE TRIMER OF YABJ. | |
-Components
| #1: Protein | Mass: 13536.371 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | ChemComp-HG / |   Mass: 200.590 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Hg Mass: 200.590 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Hg#3: Chemical | ChemComp-ACY /   Mass: 60.052 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H4O2 Mass: 60.052 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H4O2#4: Chemical | 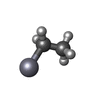  Mass: 229.651 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H5Hg Mass: 229.651 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H5Hg#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 474 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 474 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.04 Å3/Da / Density % sol: 40 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.6 Details: SODIUM PHOSPHATE, PEG 4000, AMMONIUM SULPHATE, SODIUM ACETATE., pH 4.60 | ||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / pH: 7.4 | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-D / Wavelength: 1.0095 / Beamline: 14-BM-D / Wavelength: 1.0095 |
| Detector | Type: ADSC QUANTUM 1 / Detector: CCD / Date: Jul 11, 1998 |
| Radiation | Protocol: MULTIPLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0095 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→30 Å / Num. obs: 309290 / % possible obs: 97.9 % / Observed criterion σ(I): 1 / Redundancy: 8.7 % / Biso Wilson estimate: 18.2 Å2 / Rmerge(I) obs: 0.053 / Net I/σ(I): 22.9 |
| Reflection shell | Resolution: 1.7→1.76 Å / Redundancy: 4 % / Rmerge(I) obs: 0.086 / Mean I/σ(I) obs: 8.6 / % possible all: 90.7 |
| Reflection | *PLUS Highest resolution: 1.7 Å / Lowest resolution: 30 Å / Num. obs: 35377 / Observed criterion σ(I): 1 / Redundancy: 8.7 % |
| Reflection shell | *PLUS % possible obs: 95.6 % / Redundancy: 4 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: HG MAD / Resolution: 1.7→30 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 2121779.35 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: REFINEMENT TARGET EQUALS MAXIMUM LIKELIHOOD TARGET USING AMPLITUDES AND PHASE PROBABILITY DISTRIBUTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 86.61 Å2 / ksol: 0.456 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.4 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: NONE FOR FINAL REFINEMENT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.7→1.76 Å / Rfactor Rfree error: 0.019 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 5.1 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 21.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.234 / % reflection Rfree: 4.6 % / Rfactor Rwork: 0.195 |
