 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pk5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the orphan nuclear receptor LRH-1 | ||||||
 Components Components | Orphan nuclear receptor NR5A2 | ||||||
 Keywords Keywords | GENE REGULATION / nuclear receptor / ligand-binding domain / LRH-1 | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of glucocorticoid biosynthetic process / zygotic genome activation / positive regulation of tendon cell differentiation / SUMOylation of intracellular receptors / morula formation / primary ovarian follicle growth / Nuclear Receptor transcription pathway / pancreas morphogenesis / inner cell mass cell differentiation / acinar cell differentiation ...positive regulation of glucocorticoid biosynthetic process / zygotic genome activation / positive regulation of tendon cell differentiation / SUMOylation of intracellular receptors / morula formation / primary ovarian follicle growth / Nuclear Receptor transcription pathway / pancreas morphogenesis / inner cell mass cell differentiation / acinar cell differentiation / Sertoli cell development / positive regulation of T cell anergy / positive regulation of stem cell differentiation / embryonic cleavage / bile acid metabolic process / exocrine pancreas development / cartilage development / negative regulation of chondrocyte differentiation / calcineurin-mediated signaling / somatic stem cell population maintenance / positive regulation of viral genome replication / neurogenesis / positive regulation of T cell proliferation / cholesterol homeostasis / cellular response to leukemia inhibitory factor / transcription coregulator binding / phospholipid binding / positive regulation of T cell activation / negative regulation of inflammatory response / RNA polymerase II transcription regulator complex / nuclear receptor activity / regulation of cell population proliferation / chromosome / DNA-binding transcription activator activity, RNA polymerase II-specific / spermatogenesis / RNA polymerase II cis-regulatory region sequence-specific DNA binding / chromatin remodeling / DNA-binding transcription factor activity / chromatin binding / regulation of DNA-templated transcription / positive regulation of DNA-templated transcription / positive regulation of transcription by RNA polymerase II / DNA binding / zinc ion binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Sablin, E.P. / Krylova, I.N. / Fletterick, R.J. / Ingraham, H.A. | ||||||
 Citation Citation | Journal: Mol.Cell / Year: 2003 Title: Structural basis for ligand-independent activation of the orphan nuclear receptor LRH-1 Authors: Sablin, E.P. / Krylova, I.N. / Fletterick, R.J. / Ingraham, H.A. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE Author maintains residue I525 in sequence database should be L525 |
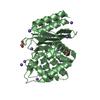
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pk5.cif.gz | 110.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pk5.ent.gz | 85.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1pk5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1pk5_validation.pdf.gz | 439.3 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1pk5_full_validation.pdf.gz | 460 KB | Display | |
| Data in XML | 1pk5_validation.xml.gz | 22.4 KB | Display | |
| Data in CIF | 1pk5_validation.cif.gz | 30.9 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pk/1pk5ftp://data.pdbj.org/pub/pdb/validation_reports/pk/1pk5 | HTTPS FTP |
-Related structure data
| Related structure data |  1fbyS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 28731.969 Da / Num. of mol.: 2 / Fragment: LRH-1 LBD Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 158 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 158 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.1 Å3/Da / Density % sol: 41 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 288 K / Method: vapor diffusion, hanging drop / pH: 8.8 Details: PEG 4000, glycerol, TRIS, isopropanol, pH 8.8, VAPOR DIFFUSION, HANGING DROP, temperature 288K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 15 ℃ / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.3.1 / Wavelength: 1.1 Å / Beamline: 8.3.1 / Wavelength: 1.1 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Sep 26, 2002 / Details: KOHZU: DOUBLE CRYSTAL SI(III) |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→25 Å / Num. all: 18102 / Num. obs: 17939 / % possible obs: 99.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 6 % / Biso Wilson estimate: 45.9 Å2 / Rmerge(I) obs: 0.067 / Rsym value: 0.067 / Net I/σ(I): 24.4 |
| Reflection shell | Resolution: 2.4→2.55 Å / Redundancy: 4 % / Rmerge(I) obs: 0.169 / Mean I/σ(I) obs: 5.7 / Rsym value: 0.169 / % possible all: 97.1 |
| Reflection | *PLUS Highest resolution: 2.4 Å / Num. obs: 17954 / % possible obs: 98.2 % |
| Reflection shell | *PLUS Highest resolution: 2.4 Å / Lowest resolution: 2.5 Å / % possible obs: 88.8 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: RXR ALPHA (PDB ID 1FBY) Resolution: 2.4→24.55 Å / Rfactor Rfree error: 0.009 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 56.3806 Å2 / ksol: 0.355359 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 48.2 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error free: 0.33 Å / Luzzati sigma a free: 0.37 Å | ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→24.55 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Software | *PLUS Classification: refinement | ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / % reflection Rfree: 5 % / Rfactor Rfree: 0.231 / Rfactor Rwork: 0.213 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
