#241 - 2020年1月 20年の分子を振り返って (Twenty Years of Molecules) 類似性 (1)
-
集合体
登録構造単位
C: 5'-D(*TP*GP*CP*TP*TP*AP*TP*CP*AP*AP*TP*TP*TP*GP*TP*TP*GP*CP*AP*CP*C)-3' D: 5'-D(*TP*GP*CP*TP*TP*AP*TP*CP*AP*AP*TP*TP*TP*GP*TP*TP*GP*CP*AP*CP*C)-3' A: DNA-binding protein HU B: DNA-binding protein HU
解像度: 1.9→25 Å / Cor.coef. Fo:Fc: 0.929 / Cor.coef. Fo:Fc free: 0.905 / SU B: 7.985 / SU ML: 0.21 / TLS residual ADP flag: LIKELY RESIDUAL / 交差検証法: THROUGHOUT / σ(F): 0 / ESU R: 0.27 / ESU R Free: 0.224 詳細: Data are anisotropic with limits 1.9 X 2.5 x 2.0. Data were truncated to an ellipsoid and reflections with an average (I/sigI) ratio less than 2 were removed. The completeness above is ...詳細: Data are anisotropic with limits 1.9 X 2.5 x 2.0. Data were truncated to an ellipsoid and reflections with an average (I/sigI) ratio less than 2 were removed. The completeness above is underestimated. When truncation is factored in, data in refinement are 91% complete. The following residues in chain A and B have some sidechain atoms with 0.00 occupancy: A3, A12, A13, A18, A19, A34, A45, A59, B3, B12, B18, B59, B67, B83, B84.
Rfactor
反射数
%反射
Selection details
Rfree
0.28788
941
4.8 %
RANDOM
Rwork
0.24415
-
-
-
obs
0.24637
18562
68.62 %
-
all
-
28420
-
-
溶媒の処理
イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.4 Å / 溶媒モデル: BABINET MODEL WITH MASK
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Anabaena sp. (バクテリア)
Anabaena sp. (バクテリア) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード






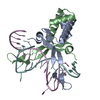






 PDBj
PDBj






































 集合体
集合体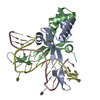
 分子量: 18.015 Da / 分子数: 196 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 196 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 14-BM-C / 波長: 1 Å
/ ビームライン: 14-BM-C / 波長: 1 Å 解析
解析