| 登録情報 | データベース: PDB / ID: 1om1
|
|---|
| タイトル | Crystal structure of maize CK2 alpha in complex with IQA |
|---|
 要素 要素 | Casein kinase II, alpha chain |
|---|
 キーワード キーワード | TRANSFERASE / CK2 / IQA / inhibitor / Zea mays |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
protein kinase CK2 complex / non-specific serine/threonine protein kinase / regulation of cell cycle / protein serine kinase activity / protein serine/threonine kinase activity / DNA damage response / ATP binding / nucleus / cytosol類似検索 - 分子機能 Casein Kinase 2, subunit alpha / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site ...Casein Kinase 2, subunit alpha / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha Beta類似検索 - ドメイン・相同性 |
|---|
| 生物種 |   Zea mays (トウモロコシ) Zea mays (トウモロコシ) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.68 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.68 Å |
|---|
 データ登録者 データ登録者 | Battistutta, R. / De Moliner, E. / Zanotti, G. |
|---|
 引用 引用 | ジャーナル: Biochem.J. / 年: 2003
タイトル: Biochemical and three-dimensional-structural study of the specific inhibition of protein kinase CK2 by [5-oxo-5,6-dihydroindolo-(1,2-a)quinazolin-7-yl]acetic acid (IQA).
著者: Sarno, S. / de Moliner, E. / Ruzzene, M. / Pagano, M.A. / Battistutta, R. / Bain, J. / Fabbro, D. / Schoepfer, J. / Elliott, M. / Furet, P. / Meggio, F. / Zanotti, G. / Pinna, L.A. |
|---|
| 履歴 | | 登録 | 2003年2月24日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2004年2月24日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2008年4月29日 | Group: Version format compliance |
|---|
| 改定 1.2 | 2011年7月13日 | Group: Version format compliance |
|---|
| 改定 1.3 | 2017年10月11日 | Group: Refinement description / カテゴリ: software / Item: _software.classification / _software.name |
|---|
| 改定 1.4 | 2024年2月14日 | Group: Data collection / Database references / Derived calculations
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード
















 PDBj
PDBj 集合体
集合体
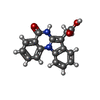
 分子量: 292.289 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C17H12N2O3
分子量: 292.289 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C17H12N2O3 分子量: 18.015 Da / 分子数: 252 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 252 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 5.2R / 波長: 1.2 Å
/ ビームライン: 5.2R / 波長: 1.2 Å 解析
解析