 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1olq: The Trichoderma reesei cel12a P201C mutant, structure at 1.7 A re... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1olq | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | The Trichoderma reesei cel12a P201C mutant, structure at 1.7 A resolution | |||||||||
 Components Components | ENDO-BETA-1,4-GLUCANASE | |||||||||
 Keywords Keywords | HYDROLASE / CELLULASE / CELLULOSE DEGRADATION / ENDOGLUCANASE / GLYCOSYL HYDROLASE / GH FAMILY 12 | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  TRICHODERMA REESEI (fungus) TRICHODERMA REESEI (fungus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | |||||||||
 Authors Authors | Sandgren, M. / Gualfetti, P.J. / Shaw, A. / Gross, L.S. / Saldajeno, M. / Berglund, G.I. / Jones, T.A. / Mitchinson, C. | |||||||||
 Citation Citation | Journal: Protein Sci. / Year: 2003 Title: The Humicola Grisea Cel12A Enzyme Structure at 1.2 A Resolution and the Impact of its Free Cysteine Residues on Thermal Stability Authors: Sandgren, M. / Gualfetti, P.J. / Paech, C. / Paech, S. / Shaw, A. / Gross, L.S. / Saldajeno, M. / Berglund, G.I. / Jones, T.A. / Mitchinson, C. | |||||||||
| History |
| |||||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1olq.cif.gz | 99.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1olq.ent.gz | 76.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1olq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ol/1olqftp://data.pdbj.org/pub/pdb/validation_reports/ol/1olq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1olrC  1h8vS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 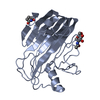
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23479.369 Da / Num. of mol.: 2 / Fragment: RESIDUES 17-234 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) TRICHODERMA REESEI (fungus) / Production host: #2: Sugar |   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 287 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 287 / Source method: isolated from a natural source / Formula: H2OCompound details | CHAIN A, B ENGINEERED | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 40 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6 / Details: pH 6.00 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20-24 ℃ / pH: 6 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.93 / Beamline: ID14-1 / Wavelength: 0.93 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Dec 3, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.93 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→20 Å / Num. obs: 40983 / % possible obs: 99.9 % / Observed criterion σ(I): 3 / Redundancy: 4.3 % / Rmerge(I) obs: 0.074 / Net I/σ(I): 18.3 |
| Reflection shell | Resolution: 1.7→1.73 Å / Redundancy: 99.6 % / Rmerge(I) obs: 0.39 / Mean I/σ(I) obs: 2.8 |
| Reflection | *PLUS Highest resolution: 1.7 Å / Lowest resolution: 20 Å / Num. obs: 42832 / % possible obs: 99.8 % / Redundancy: 4.3 % / Num. measured all: 184371 / Rmerge(I) obs: 0.074 |
| Reflection shell | *PLUS % possible obs: 99.6 % / Rmerge(I) obs: 0.39 / Mean I/σ(I) obs: 2.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1H8V Resolution: 1.7→20 Å / SU B: 4.26607 / SU ML: 0.14491 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.24664 / ESU R Free: 0.18176 Details: BABINET'S PRINCIPLE FOR SCALING HAS BEEN USED. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIO BULK SOLVENT MODELLING. PARAMETERS FOR MASK CALCULATION THE FOLLOWING REFINEMENT PARAMETERS ARE ...Details: BABINET'S PRINCIPLE FOR SCALING HAS BEEN USED. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIO BULK SOLVENT MODELLING. PARAMETERS FOR MASK CALCULATION THE FOLLOWING REFINEMENT PARAMETERS ARE NOT REPRESENTED IN THE ABOVE TEMPLATE FOR REFMAC
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 13.208 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.7 Å / Num. reflection obs: 40981 / Rfactor Rfree: 0.251 / Rfactor Rwork: 0.209 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
