 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nki: CRYSTAL STRUCTURE OF THE FOSFOMYCIN RESISTANCE PROTEIN A (FOSA) C... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nki | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE FOSFOMYCIN RESISTANCE PROTEIN A (FOSA) CONTAINING BOUND PHOSPHONOFORMATE | ||||||
 Components Components | probable fosfomycin resistance protein | ||||||
 Keywords Keywords | TRANSFERASE / POTASSIUM BINDING LOOP / MANGANESE BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationglutathione transferase / glutathione transferase activity / response to antibiotic / metal ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO / Resolution: 0.95 Å X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO / Resolution: 0.95 Å | ||||||
 Authors Authors | Rife, C.L. / Pharris, R.E. / Newcomer, M.E. / Armstrong, R.N. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2004 Title: Phosphonoformate: a minimal transition state analogue inhibitor of the fosfomycin resistance protein, FosA. Authors: Rigsby, R.E. / Rife, C.L. / Fillgrove, K.L. / Newcomer, M.E. / Armstrong, R.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nki.cif.gz | 140.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nki.ent.gz | 108.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1nki.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nk/1nkiftp://data.pdbj.org/pub/pdb/validation_reports/nk/1nki | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1nnrC  1lqpS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | a dimer of chains A and B is the complete biological assembly |
-Components
| #1: Protein | Mass: 15143.006 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria) / Strain: PAO1 / Plasmid: pet-20 / Species (production host): Escherichia coli / Production host: #2: Chemical |   Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K#3: Chemical |   Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn#4: Chemical | 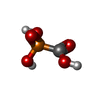  Mass: 126.005 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH3O5P / Comment: medication, antivirus, inhibitor*YM Mass: 126.005 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH3O5P / Comment: medication, antivirus, inhibitor*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 419 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 419 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.94 Å3/Da / Density % sol: 35.94 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7 Details: PENTAERYTHRITOL PROPOXYLATE, MANGANESE CHLORIDE, POTASSIUM PHOSPHATE, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
| Crystal grow | *PLUS Method: unknown / Details: Rife, C.L., (2002) J.Am.Chem.Soc., 124, 11001. |
-Data collection
| Diffraction | Mean temperature: 113 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-C / Wavelength: 0.9 Å / Beamline: 14-BM-C / Wavelength: 0.9 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jun 20, 2002 / Details: Bent conical Si-mirror |
| Radiation | Monochromator: Bent Ge(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 0.95→30 Å / Num. all: 178057 / Num. obs: 154452 / % possible obs: 91.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 8.4 % / Rmerge(I) obs: 0.058 / Net I/σ(I): 26 |
| Reflection shell | Resolution: 0.95→0.98 Å / Rmerge(I) obs: 0.358 / Mean I/σ(I) obs: 2.28 / % possible all: 46.8 |
| Reflection shell | *PLUS % possible obs: 46.8 % / Mean I/σ(I) obs: 2.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: AB INITIO Starting model: PDB ENTRY 1LQP Resolution: 0.95→30 Å / Num. parameters: 23457 / Num. restraintsaints: 28614 / Cross valid method: FREE R / σ(F): 0 / σ(I): -3 / Stereochemistry target values: ENGH AND HUBER Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 4 PERCENT
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 6 / Occupancy sum hydrogen: 1992 / Occupancy sum non hydrogen: 2562.95 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.95→30 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 30 Å / Rfactor Rfree: 0.161 / Rfactor Rwork: 0.132 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
