 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1naq | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | Crystal structure of CUTA1 from E.coli at 1.7 A resolution | ||||||
 要素 要素 | Periplasmic divalent cation tolerance protein cutA | ||||||
 キーワード キーワード | ELECTRON TRANSPORT / CUTA / copper resistance / Structural Proteomics in Europe / SPINE / Structural Genomics | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報response to copper ion / copper ion binding / protein-containing complex / metal ion binding / cytoplasm 類似検索 - 分子機能 | ||||||
| 生物種 |  | ||||||
| 手法 |  X線回折 / シンクロトロン / 多波長異常分散 / 解像度: 1.7 Å X線回折 / シンクロトロン / 多波長異常分散 / 解像度: 1.7 Å | ||||||
 データ登録者 データ登録者 | Calderone, V. / Mangani, S. / Benvenuti, M. / Viezzoli, M.S. / Banci, L. / Bertini, I. / Structural Proteomics in Europe (SPINE) | ||||||
 引用 引用 | ジャーナル: J.Biol.Chem. / 年: 2003 タイトル: The evolutionarily conserved trimeric structure of CutA1 proteins suggests a role in signal transduction. 著者: Arnesano, F. / Banci, L. / Benvenuti, M. / Bertini, I. / Calderone, V. / Mangani, S. / Viezzoli, M.S. #1: ジャーナル: To be Publishedタイトル: STRUCTURE OF PROTEIN TM1056, CUTA 著者: SAVCHENKO, A. / ZHANG, R. / JOACHIMIAK, A. / EDWARDS, A. / AKARINA, T. | ||||||
| 履歴 |
| ||||||
| Remark 600 | HETEROGEN P-HYDROXYMERCURIBENZOIC ACID HAS BEEN ADDED TO THE PROTEIN PRIOR TO CRYSTALLISATION. IT ...HETEROGEN P-HYDROXYMERCURIBENZOIC ACID HAS BEEN ADDED TO THE PROTEIN PRIOR TO CRYSTALLISATION. IT REACTS WITH THE -SH OF FREE CYSTEINS AND, BY THE ELIMINATION OF ONE WATER MOLECULE, FORMS A COVALENT BOND BETWEEN THE S OF THE CYS AND HG WHICH IS THEN A GOOD CANDIDATE TO PERFORM A MAD EXPERIMENT. |
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1naq.cif.gz | 143.5 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1naq.ent.gz | 114.3 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1naq.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/na/1naqftp://data.pdbj.org/pub/pdb/validation_reports/na/1naq | HTTPS FTP |
|---|
-関連構造データ












-リンク
 PDBj
PDBj













- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 |
| ||||||||
| 単位格子 |
| ||||||||
| 詳細 | The biological functional unit is a trimer; the asymmetric unit is made of two trimers. |
-要素
| #1: タンパク質 | 分子量: 12342.025 Da / 分子数: 6 / 由来タイプ: 組換発現 / 由来: (組換発現) #2: 化合物 | ChemComp-HG /   分子量: 200.590 Da / 分子数: 8 / 由来タイプ: 合成 / 式: Hg 分子量: 200.590 Da / 分子数: 8 / 由来タイプ: 合成 / 式: Hg#3: 化合物 | ChemComp-MBO / 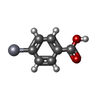  分子量: 321.703 Da / 分子数: 10 / 由来タイプ: 合成 / 式: C7H5HgO2 分子量: 321.703 Da / 分子数: 10 / 由来タイプ: 合成 / 式: C7H5HgO2#4: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 343 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 343 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.07 Å3/Da / 溶媒含有率: 40.56 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 293 K / 手法: 蒸気拡散法, シッティングドロップ法 / pH: 7.5 詳細: 0.1M Na HEPES, 2M Ammonium sulphate, 2% PEG 400, 2 mM 4-(hydroxymercuri)benzoic acid, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 293K | ||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 20 ℃ / 手法: 蒸気拡散法 | ||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: EMBL/DESY, HAMBURG  / ビームライン: BW7A / 波長: 1.005231, 1.00870, 0.93200 / ビームライン: BW7A / 波長: 1.005231, 1.00870, 0.93200 | ||||||||||||
| 検出器 | タイプ: MARRESEARCH / 検出器: CCD / 日付: 2002年8月14日 | ||||||||||||
| 放射 | モノクロメーター: Double crystal focussing monochromator プロトコル: MAD / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray | ||||||||||||
| 放射波長 |
| ||||||||||||
| 反射 | 解像度: 1.7→37.5 Å / Num. all: 65739 / Num. obs: 65739 / % possible obs: 97.2 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / 冗長度: 9.7 % / Biso Wilson estimate: 18.95 Å2 / Rmerge(I) obs: 0.091 / Rsym value: 0.091 / Net I/σ(I): 5.7 | ||||||||||||
| 反射 シェル | 解像度: 1.7→1.79 Å / 冗長度: 9.6 % / Rmerge(I) obs: 0.619 / Mean I/σ(I) obs: 1.1 / Num. unique all: 9337 / Rsym value: 0.619 / % possible all: 96.3 | ||||||||||||
| 反射 | *PLUS 最低解像度: 40 Å / Num. measured all: 643844 | ||||||||||||
| 反射 シェル | *PLUS % possible obs: 96.3 % / Num. unique obs: 9337 / Num. measured obs: 90452 / Rmerge(I) obs: 0.519 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多波長異常分散 / 解像度: 1.7→19.96 Å / Cor.coef. Fo:Fc: 0.938 / Cor.coef. Fo:Fc free: 0.903 / SU B: 3.338 / SU ML: 0.11 / 交差検証法: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.164 / ESU R Free: 0.152 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD 詳細: At the N-terminus of all the six molecules present in the asymetric unit there are about 6-8 residues for which it's not possible to see a clear density. Among the densities belonging to each ...詳細: At the N-terminus of all the six molecules present in the asymetric unit there are about 6-8 residues for which it's not possible to see a clear density. Among the densities belonging to each asymmetric unit it's possible to see some extra density which could account for the presence of some crystalline PEG fragments.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.4 Å / 溶媒モデル: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 20.738 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati sigma a obs: 0.055714 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.7→19.96 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.7→1.79 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS 最高解像度: 1.7 Å / 最低解像度: 20 Å / Rfactor Rfree: 0.255 / Rfactor Rwork: 0.203 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
