 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1mq0 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Crystal Structure of Human Cytidine Deaminase | |||||||||
 要素 要素 | Cytidine Deaminase | |||||||||
 キーワード キーワード | HYDROLASE / human / enzyme / cytidine deaminase / amine hydrolase / inhibitor / diazepinone / leukemia / chemotherapy / anticancer / drug / phi-phi interaction / edge-to-face interaction / protein | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報negative regulation of nucleotide metabolic process / pyrimidine-containing compound salvage / cytidine deaminase / Pyrimidine salvage / : / cellular response to external biotic stimulus / cytidine deaminase activity / UMP salvage / nucleoside binding / cytosine metabolic process ...negative regulation of nucleotide metabolic process / pyrimidine-containing compound salvage / cytidine deaminase / Pyrimidine salvage / : / cellular response to external biotic stimulus / cytidine deaminase activity / UMP salvage / nucleoside binding / cytosine metabolic process / response to cycloheximide / negative regulation of cell growth / tertiary granule lumen / secretory granule lumen / ficolin-1-rich granule lumen / cell surface receptor signaling pathway / Neutrophil degranulation / protein homodimerization activity / extracellular region / zinc ion binding / identical protein binding / cytosol 類似検索 - 分子機能 | |||||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | |||||||||
| 手法 |  X線回折 / 分子置換 / 解像度: 2.4 Å X線回折 / 分子置換 / 解像度: 2.4 Å | |||||||||
 データ登録者 データ登録者 | Chung, S.J. / Fromme, J.C. / Verdine, G.L. | |||||||||
 引用 引用 | ジャーナル: J.Med.Chem. / 年: 2005 タイトル: Structure of human cytidine deaminase bound to a potent inhibitor 著者: Chung, S.J. / Fromme, J.C. / Verdine, G.L. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1mq0.cif.gz | 65 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1mq0.ent.gz | 46.7 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1mq0.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1mq0_validation.pdf.gz | 461.2 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1mq0_full_validation.pdf.gz | 468.5 KB | 表示 | |
| XML形式データ | 1mq0_validation.xml.gz | 15.5 KB | 表示 | |
| CIF形式データ | 1mq0_validation.cif.gz | 19.9 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/mq/1mq0ftp://data.pdbj.org/pub/pdb/validation_reports/mq/1mq0 | HTTPS FTP |
-関連構造データ
| 関連構造データ |  1jtkS S: 精密化の開始モデル |
|---|---|
| 類似構造データ |









-リンク
 PDBj
PDBj- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 単位格子 |
| ||||||||
| 詳細 | The biological unit is a tetramer, and there is a dimer in the asymmetric unit. The other dimer of the tetramer can be obtained by the following symmetry operation: (-X, -Y,Z) dx=1 dy=3 dz=0 |
-要素
| #1: タンパク質 | 分子量: 15510.718 Da / 分子数: 2 / 変異: K27Q / 由来タイプ: 組換発現 / 由来: (組換発現) Homo sapiens (ヒト) / 生物種 (発現宿主): Escherichia coli / 発現宿主:  #2: 化合物 |   分子量: 65.409 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Zn 分子量: 65.409 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Zn#3: 化合物 | 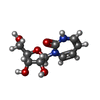  分子量: 244.244 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C10H16N2O5 分子量: 244.244 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C10H16N2O5#4: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 62 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 62 / 由来タイプ: 天然 / 式: H2OHas protein modification | N | |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.13 Å3/Da / 溶媒含有率: 42.32 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 298 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 4.6 詳細: sodium acetate, 2-methyl-2,4-pentane-diol, calcium chloride, synthetic inhibitor, pH 4.6, VAPOR DIFFUSION, HANGING DROP, temperature 298K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 4 ℃ / pH: 7.5 / 手法: 蒸気拡散法, ハンギングドロップ法 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU ULTRAX 18 / 波長: 1.5418 Å |
| 検出器 | タイプ: RIGAKU RAXIS IV / 検出器: IMAGE PLATE / 日付: 2002年7月19日 / 詳細: Osmic Blue |
| 放射 | モノクロメーター: Nickel filter / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 解像度: 2.3→47.45 Å / Num. obs: 11082 / % possible obs: 97.1 % / Observed criterion σ(I): 0 / 冗長度: 4.58 % / Biso Wilson estimate: 28.5 Å2 / Rmerge(I) obs: 0.09 / Net I/σ(I): 10.4 |
| 反射 シェル | 解像度: 2.3→2.38 Å / 冗長度: 4.56 % / Rmerge(I) obs: 0.326 / Mean I/σ(I) obs: 2.9 / Num. unique all: 1134 / % possible all: 98.1 |
| 反射 | *PLUS 最高解像度: 2.3 Å / Num. obs: 22165 / % possible obs: 97.1 % / 冗長度: 4.4 % |
| 反射 シェル | *PLUS % possible obs: 98.1 % / 冗長度: 4.5 % / Rmerge(I) obs: 0.33 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: PDB ENTRY 1JTK 解像度: 2.4→47.45 Å / Rfactor Rfree error: 0.011 / Data cutoff high absF: 210371.16 / Data cutoff high rms absF: 210371.16 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: FLAT MODEL / Bsol: 51.7547 Å2 / ksol: 0.338176 e/Å3 | ||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 38.9 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.4→47.45 Å
| ||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2.4→2.55 Å / Rfactor Rfree error: 0.037 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS % reflection Rfree: 10 % | ||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
