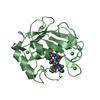
 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mmp | ||||||
|---|---|---|---|---|---|---|---|
| Title | MATRILYSIN COMPLEXED WITH CARBOXYLATE INHIBITOR | ||||||
 Components Components | GELATINASE A | ||||||
 Keywords Keywords | METALLOPROTEASE | ||||||
| Function / homology |  Function and homology information Function and homology informationmatrilysin / antibacterial peptide secretion / antibacterial peptide biosynthetic process / membrane protein intracellular domain proteolysis / Assembly of collagen fibrils and other multimeric structures / Activation of Matrix Metalloproteinases / Collagen degradation / collagen catabolic process / membrane protein ectodomain proteolysis / extracellular matrix disassembly ...matrilysin / antibacterial peptide secretion / antibacterial peptide biosynthetic process / membrane protein intracellular domain proteolysis / Assembly of collagen fibrils and other multimeric structures / Activation of Matrix Metalloproteinases / Collagen degradation / collagen catabolic process / membrane protein ectodomain proteolysis / extracellular matrix disassembly / Degradation of the extracellular matrix / extracellular matrix organization / metalloendopeptidase activity / metallopeptidase activity / regulation of cell population proliferation / extracellular matrix / endopeptidase activity / defense response to Gram-negative bacterium / Extra-nuclear estrogen signaling / defense response to Gram-positive bacterium / positive regulation of cell migration / response to xenobiotic stimulus / serine-type endopeptidase activity / proteolysis / : / extracellular exosome / extracellular region / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.3 Å X-RAY DIFFRACTION / Resolution: 2.3 Å | ||||||
 Authors Authors | Browner, M.F. / Smith, W.W. / Castelhano, A.L. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1995 Title: Matrilysin-inhibitor complexes: common themes among metalloproteases. Authors: Browner, M.F. / Smith, W.W. / Castelhano, A.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mmp.cif.gz | 81.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mmp.ent.gz | 60.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1mmp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mm/1mmpftp://data.pdbj.org/pub/pdb/validation_reports/mm/1mmp | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18741.912 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Organ: OVARY / Production host:   Cricetulus griseus (Chinese hamster) / References: UniProt: P09237, matrilysin Cricetulus griseus (Chinese hamster) / References: UniProt: P09237, matrilysin#2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#4: Chemical | 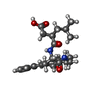  Mass: 441.563 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C25H35N3O4 Mass: 441.563 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C25H35N3O4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 89 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 89 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 52.68 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal | *PLUS Density % sol: 51.3 % | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 17-25 ℃ / pH: 6.5 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Wavelength: 1.5418 Å |
|---|---|
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 27, 1993 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Num. obs: 15769 / % possible obs: 86.2 % / Observed criterion σ(I): 2.5 / Redundancy: 5.1 % / Rmerge(I) obs: 0.09 |
| Reflection | *PLUS Highest resolution: 2.4 Å / Num. measured all: 80416 / Rmerge(I) obs: 0.09 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.3→6 Å / σ(F): 0 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
