 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lej: NMR Structure of a 1:1 Complex of Polyamide (Im-Py-Beta-Im-Beta-I... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lej | ||||||
|---|---|---|---|---|---|---|---|
| Title | NMR Structure of a 1:1 Complex of Polyamide (Im-Py-Beta-Im-Beta-Im-Py-Beta-Dp) with the Tridecamer DNA Duplex 5'-CCAAAGAGAAGCG-3' | ||||||
 Components Components |
| ||||||
 Keywords Keywords | DNA / POLYAMIDE / PURINE TRACT / BETA ALANINE / MINOR GROOVE | ||||||
| Function / homology | Chem-P11 / DNA / DNA (> 10) Function and homology information Function and homology information | ||||||
| Method | SOLUTION NMR / restrained molecular dynamics, simulated annealing, matrix relaxation, NOE-derived distance restraints. | ||||||
 Authors Authors | Urbach, A.R. / Love, J.J. / Ross, S.A. / Dervan, P.B. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2002 Title: Structure of a beta-alanine-linked polyamide bound to a full helical turn of purine tract DNA in the 1:1 motif. Authors: Urbach, A.R. / Love, J.J. / Ross, S.A. / Dervan, P.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lej.cif.gz | 208.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lej.ent.gz | 168.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1lej.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/le/1lejftp://data.pdbj.org/pub/pdb/validation_reports/le/1lej | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|




-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 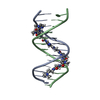
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: DNA chain | Mass: 4018.652 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: This sequence was designed and synthesized for this study. |
|---|---|
| #2: DNA chain | Mass: 3924.544 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: This sequence was designed and synthesized for this study. |
| #3: Chemical | ChemComp-P11 / 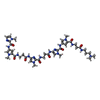  Mass: 914.992 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C41H56N17O8 Mass: 914.992 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C41H56N17O8 |
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR |
|---|---|
| NMR experiment | Type: 2D NOESY |
| NMR details | Text: This structure was determined using standard 2D homonuclear techniques. |
- Sample preparation
Sample preparation
| Details | Contents: 3.67 millimolar polyamide-DNA complex, 10 millimolar sodium phosphate, pH 7.0, 90% H2O, 10% D2O Solvent system: 90% H2O/10% D2O |
|---|---|
| Sample conditions | Ionic strength: 10 Millimolar / pH: 7 / Pressure: ambient / Temperature: 298 K |
| Crystal grow | *PLUS Method: other / Details: NMR |
-NMR measurement
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| NMR spectrometer | Type: Varian INOVA / Manufacturer: Varian / Model: INOVA / Field strength: 600 MHz |
- Processing
Processing
| NMR software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method: restrained molecular dynamics, simulated annealing, matrix relaxation, NOE-derived distance restraints. Software ordinal: 1 Details: The structure calculations used 548 distance restraints. 508 were NOE-derived, and 40 were for Watson-Crick hydrogen bonds. | ||||||||||||||||||||||||||||
| NMR representative | Selection criteria: fewest violations | ||||||||||||||||||||||||||||
| NMR ensemble | Conformer selection criteria: structures with the least restraint violations Conformers calculated total number: 40 / Conformers submitted total number: 11 |
 NMRPipe
NMRPipe