 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lb7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | IGF-F1-1, A PEPTIDE ANTAGONIST OF IGF-1 | ||||||
 Components Components | IGF-1 ANTAGONIST F1-1 | ||||||
 Keywords Keywords | DE NOVO PROTEIN / loop-helix / disulfide | ||||||
| Method | SOLUTION NMR / distance geometry, molecular dynamics | ||||||
 Authors Authors | Deshayes, K. / Schaffer, M.L. / Skelton, N.J. / Nakamura, G.R. / Kadkhodayan, S. / Sidhu, S.S. | ||||||
 Citation Citation | Journal: Chem.Biol. / Year: 2002 Title: Rapid identification of small binding motifs with high-throughput phage display: discovery of peptidic antagonists of IGF-1 function. Authors: Deshayes, K. / Schaffer, M.L. / Skelton, N.J. / Nakamura, G.R. / Kadkhodayan, S. / Sidhu, S.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule:  MolmilJmol/JSmol MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lb7.cif.gz | 93.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lb7.ent.gz | 64.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1lb7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lb/1lb7ftp://data.pdbj.org/pub/pdb/validation_reports/lb/1lb7 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|





-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 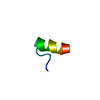
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: Protein/peptide | Mass: 1879.216 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: De novo sequence derived from phage display library |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NMR experiment |
| ||||||||||||||||||||||||
| NMR details | Text: This structure was determined using standard 2D homonuclear techniques to generate distance (ROESY) and dihederal angle (DQF-COSY, COSY-35)restraints. Additional dihedral angle restraints were ...Text: This structure was determined using standard 2D homonuclear techniques to generate distance (ROESY) and dihederal angle (DQF-COSY, COSY-35)restraints. Additional dihedral angle restraints were derived from chemical shift information using the program TALOS |
 HSQC
HSQC- Sample preparation
Sample preparation
| Details |
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample conditions | Ionic strength: 0 / pH: 5.1 / Pressure: atm / Temperature: 303 K | |||||||||
| Crystal grow | *PLUS Method: other / Details: NMR |
-NMR measurement
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Radiation wavelength | Relative weight: 1 | |||||||||||||||
| NMR spectrometer |
|
- Processing
Processing
| NMR software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method: distance geometry, molecular dynamics / Software ordinal: 1 Details: The structures are based on 91 distance restraints and 26 dihedral angle restraints. None of the final structures have violations > 0.07A or 1.0 degrees. The mean rmsd to the mean ...Details: The structures are based on 91 distance restraints and 26 dihedral angle restraints. None of the final structures have violations > 0.07A or 1.0 degrees. The mean rmsd to the mean coordinates for heavy atoms of residues Cys3 to Tyr15 is 0.31+/-0.06 A. | ||||||||||||||||
| NMR representative | Selection criteria: closest to the average,fewest violations,lowest energy,minimized average structure | ||||||||||||||||
| NMR ensemble | Conformer selection criteria: structures with the least restraint violations Conformers calculated total number: 80 / Conformers submitted total number: 20 |
