登録情報 データベース : PDB / ID : 1l2qタイトル Crystal Structure of the Methanosarcina barkeri Monomethylamine Methyltransferase (MtmB) monomethylamine methyltransferase キーワード / 機能・相同性 分子機能 ドメイン・相同性 構成要素
/ / / / / / / / / / 生物種 Methanosarcina barkeri (古細菌)手法 / / / 解像度 : 1.7 Å データ登録者 Hao, B. / Gong, W. / Ferguson, T.K. / James, C.M. / Krzycki, J.A. / Chan, M.K. ジャーナル : Science / 年 : 2002タイトル : A new UAG-encoded residue in the structure of a methanogen methyltransferase.著者 : Hao, B. / Gong, W. / Ferguson, T.K. / James, C.M. / Krzycki, J.A. / Chan, M.K. 履歴 登録 2002年2月24日 登録サイト / 処理サイト 改定 1.0 2002年6月5日 Provider / タイプ 改定 1.1 2008年4月28日 Group 改定 1.2 2011年7月13日 Group / Version format compliance改定 1.3 2014年8月13日 Group / Structure summary改定 1.4 2025年3月26日 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Structure summary カテゴリ chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_entry_details / pdbx_modification_feature / pdbx_poly_seq_scheme / struct_conn / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_poly_seq_scheme.auth_seq_num / _struct_conn.pdbx_leaving_atom_flag / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
すべて表示 表示を減らす Remark 600 HETEROGEN THE AUTHORS HAVE INDICATED THAT THE METHYL GROUP IN THE RESIDUE XPL202A(CONFORMER A) AND ... HETEROGEN THE AUTHORS HAVE INDICATED THAT THE METHYL GROUP IN THE RESIDUE XPL202A(CONFORMER A) AND PYL202A(CONFORMER B) COULD BE A METHYL (CH3), AMINE (NH2), OR HYDOXYL (OH) GROUP.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Methanosarcina barkeri (古細菌)
Methanosarcina barkeri (古細菌) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード








 PDBj
PDBj
 集合体
集合体

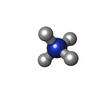
 分子量: 18.038 Da / 分子数: 1 / 由来タイプ: 合成 / 式: H4N
分子量: 18.038 Da / 分子数: 1 / 由来タイプ: 合成 / 式: H4N 分子量: 18.015 Da / 分子数: 533 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 533 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 14-BM-C / 波長: 1 Å
/ ビームライン: 14-BM-C / 波長: 1 Å 解析
解析