 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
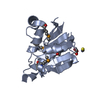
Yorodumi- PDB-1l1d: Crystal structure of the C-terminal methionine sulfoxide reductas... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1l1d | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the C-terminal methionine sulfoxide reductase domain (MsrB) of N. gonorrhoeae pilB | ||||||
 Components Components | peptide methionine sulfoxide reductase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / cacodylate complex / cys-arg-asp catalytic triad / met-R(O) reductase / MsrB | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptide-methionine (R)-S-oxide reductase / peptide-methionine (R)-S-oxide reductase activity / L-methionine (S)-S-oxide reductase activity / peptide-methionine (S)-S-oxide reductase / peptide-methionine (S)-S-oxide reductase activity / cellular response to oxidative stress / cytoplasm Similarity search - Function | ||||||
| Biological species |  Neisseria gonorrhoeae (bacteria) Neisseria gonorrhoeae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.85 Å | ||||||
 Authors Authors | Lowther, W.T. / Weissbach, H. / Etienne, F. / Brot, N. / Matthews, B.W. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 2002 Title: The mirrored methionine sulfoxide reductases of Neisseria gonorrhoeae pilB. Authors: Lowther, W.T. / Weissbach, H. / Etienne, F. / Brot, N. / Matthews, B.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1l1d.cif.gz | 72.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1l1d.ent.gz | 54.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1l1d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l1/1l1dftp://data.pdbj.org/pub/pdb/validation_reports/l1/1l1d | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 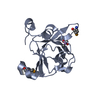
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 17020.541 Da / Num. of mol.: 2 / Fragment: MsrB domain, C-terminal domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Neisseria gonorrhoeae (bacteria) / Gene: pilB / Plasmid: pet28b / Species (production host): Escherichia coli / Production host: References: UniProt: P14930, EC: 1.8.4.13, L-methionine (R)-S-oxide reductase #2: Chemical | 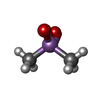  Mass: 136.989 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6AsO2 Mass: 136.989 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6AsO2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 124 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 124 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.13 Å3/Da / Density % sol: 41.8 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: PEG 4000, Tris, cacodylate, glycerol, pH 8.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 103 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.2 / Wavelength: 0.9792, 0.9793, 0.9611 / Beamline: 5.0.2 / Wavelength: 0.9792, 0.9793, 0.9611 | ||||||||||||
| Detector | Type: ADSC QUANTUM / Detector: CCD / Date: Jul 21, 2001 | ||||||||||||
| Radiation | Monochromator: Double crystal / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 1.85→27.2 Å / Num. obs: 22584 / % possible obs: 89.5 % / Observed criterion σ(I): -3 / Biso Wilson estimate: 27.9 Å2 / Rmerge(I) obs: 0.066 / Net I/σ(I): 22.3 | ||||||||||||
| Reflection shell | Resolution: 1.85→1.92 Å / Rmerge(I) obs: 0.245 / Mean I/σ(I) obs: 4.6 / % possible all: 89 | ||||||||||||
| Reflection | *PLUS Num. measured all: 98562 / Rmerge(I) obs: 0.066 | ||||||||||||
| Reflection shell | *PLUS % possible obs: 89.1 % / Rmerge(I) obs: 0.245 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.85→27.2 Å / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: THE STRUCTURE FACTORS REPRESENT THE OPTIMIZED REFERENCE STRUCTURE FACTORS (FPSHA) AS DETERMINED BY SHARP. THE REFERENCE STRUCTURE FACTOR FOR A GIVEN REFLECTION IS DEFINED AS THE AVERAGE OF ...Details: THE STRUCTURE FACTORS REPRESENT THE OPTIMIZED REFERENCE STRUCTURE FACTORS (FPSHA) AS DETERMINED BY SHARP. THE REFERENCE STRUCTURE FACTOR FOR A GIVEN REFLECTION IS DEFINED AS THE AVERAGE OF THE STRUCTURE FACTORS FROM A THREE-WAVELENGTH MAD DATASET WHERE THE HEAVY-ATOM STRUCTURE FACTOR HAS BEEN SUBTRACTED.
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 20.7 Å2 | ||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.236 Å | ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→27.2 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 10 % / Rfactor obs: 0.207 / Rfactor Rfree: 0.237 / Rfactor Rwork: 0.207 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS |
