 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kv7: Crystal Structure of CueO, a multi-copper oxidase from E. coli in... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kv7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of CueO, a multi-copper oxidase from E. coli involved in copper homeostasis | ||||||
 Components Components | PROBABLE BLUE-COPPER PROTEIN YACK | ||||||
 Keywords Keywords | OXIDOREDUCTASE / multi-copper oxidase / T1 (blue) copper | ||||||
| Function / homology |  Function and homology information Function and homology informationoxidoreductase activity, acting on metal ions, oxygen as acceptor / cuproxidase / oxidoreductase activity, acting on metal ions / detoxification of copper ion / oxidoreductase activity, acting on diphenols and related substances as donors, oxygen as acceptor / response to copper ion / ferroxidase activity / outer membrane-bounded periplasmic space / periplasmic space / copper ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.4 Å | ||||||
 Authors Authors | Roberts, S.A. / Weichsel, A. / Grass, G. / Thakali, K. / Hazzard, J.T. / Tollin, G. / Rensing, C. / Montfort, W.R. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2002 Title: Crystal structure and electron transfer kinetics of CueO, a multicopper oxidase required for copper homeostasis in Escherichia coli. Authors: Roberts, S.A. / Weichsel, A. / Grass, G. / Thakali, K. / Hazzard, J.T. / Tollin, G. / Rensing, C. / Montfort, W.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kv7.cif.gz | 115.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kv7.ent.gz | 86.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1kv7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kv/1kv7ftp://data.pdbj.org/pub/pdb/validation_reports/kv/1kv7 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 53483.285 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||||
|---|---|---|---|---|---|
| #2: Chemical |   Mass: 63.546 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cu Mass: 63.546 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cu#3: Chemical | ChemComp-C2O / | 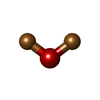  Mass: 143.091 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cu2O Mass: 143.091 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cu2O#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 507 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 507 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.18 Å3/Da / Density % sol: 43.58 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 4.6 Details: 20% PEG, 200 mM ammonium acetate, 100 mM sodium acetate, pH 4.6, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-2 / Wavelength: 1.000, 1.3772, 1.1922, 1.3804 / Beamline: BL9-2 / Wavelength: 1.000, 1.3772, 1.1922, 1.3804 | |||||||||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jun 28, 2001 | |||||||||||||||
| Radiation | Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 1.4→26 Å / Num. all: 88158 / Num. obs: 88158 / % possible obs: 97 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Rsym value: 0.064 / Net I/σ(I): 7 | |||||||||||||||
| Reflection shell | Resolution: 1.4→1.49 Å / Mean I/σ(I) obs: 2.3 / Rsym value: 0.28 / % possible all: 87 | |||||||||||||||
| Reflection | *PLUS Highest resolution: 1.4 Å / Num. obs: 88826 / % possible obs: 98.5 % / Num. measured all: 733346 / Rmerge(I) obs: 0.064 | |||||||||||||||
| Reflection shell | *PLUS % possible obs: 87.1 % / Rmerge(I) obs: 0.28 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.4→26 Å / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: Missing from the model are residues 1-30 at the N-terminus (1-28 are a presumably cleaved signal peptide, 29-30 are not visible in the electron density map), and residues 380-402 which are ...Details: Missing from the model are residues 1-30 at the N-terminus (1-28 are a presumably cleaved signal peptide, 29-30 are not visible in the electron density map), and residues 380-402 which are not visible in the electron density map and, presumably, disordered.
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.4→26 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | ||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 30 Å / % reflection Rfree: 5 % / Rfactor all: 0.19 / Rfactor obs: 0.185 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
