 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kme: CRYSTAL STRUCTURE OF BACTERIORHODOPSIN CRYSTALLIZED FROM BICELLES -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kme | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF BACTERIORHODOPSIN CRYSTALLIZED FROM BICELLES | ||||||
 Components Components | Bacteriorhodopsin | ||||||
 Keywords Keywords | MEMBRANE PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationlight-driven active monoatomic ion transmembrane transporter activity / photoreceptor activity / phototransduction / monoatomic ion channel activity / proton transmembrane transport / plasma membrane Similarity search - Function | ||||||
| Biological species |  Halobacterium salinarum (Halophile) Halobacterium salinarum (Halophile) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Faham, S. / Bowie, J.U. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2002 Title: Bicelle crystallization: a new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. Authors: Faham, S. / Bowie, J.U. | ||||||
| History |
| ||||||
| Remark 600 | HETEROGEN The identification of the lipid fragment as SQU is simply a guess. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kme.cif.gz | 101.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kme.ent.gz | 78.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1kme.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/km/1kmeftp://data.pdbj.org/pub/pdb/validation_reports/km/1kme | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1c3wS S: Starting model for refinement |
|---|---|
| Similar structure data |

















-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The two molecules in the asymmetric unit are related by this rotation matrix : ( 0.99998 0.00411 -0.00372 ) ( 0.00410 -0.99999 -0.00269 ) ( -0.00373 0.00268 -0.99999 ) and this translation ( -22.61935 103.36683 34.80453 ) |
-Components
| #1: Protein | Mass: 25301.916 Da / Num. of mol.: 2 / Fragment: Residues 14-244 / Source method: isolated from a natural source / Source: (natural) Halobacterium salinarum (Halophile) / References: UniProt: P02945#2: Chemical |   Mass: 284.436 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H28O Mass: 284.436 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H28O#3: Chemical |   Mass: 380.734 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H56 Mass: 380.734 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H56#4: Sugar | 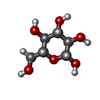  Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C6H12O6 #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 122 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 122 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.48 Å3/Da / Density % sol: 50.4 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 310 K / Method: vapor diffusion, hanging drop, bicelle method / pH: 3.5 Details: sodium phosphate, DMPC, Chapso, pH 3.5, VAPOR DIFFUSION, HANGING DROP, Bicelle Method, temperature 310.0K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 37 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X8C / Wavelength: 0.9795 Å / Beamline: X8C / Wavelength: 0.9795 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Apr 17, 2001 |
| Radiation | Monochromator: MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 2→50 Å / Num. all: 31440 / Num. obs: 31440 / % possible obs: 92.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.3 % / Biso Wilson estimate: 5 Å2 / Rmerge(I) obs: 0.102 / Net I/σ(I): 11.2 |
| Reflection shell | Resolution: 2→2.07 Å / Rmerge(I) obs: 0.26 / Mean I/σ(I) obs: 3.2 / % possible all: 62.5 |
| Reflection | *PLUS Num. measured all: 99274 |
| Reflection shell | *PLUS % possible obs: 62.5 % / Rmerge(I) obs: 0.26 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1C3W Resolution: 2→41.24 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 1159567.11 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 114.27 Å2 / ksol: 0.444285 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→41.24 Å
| ||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.021 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 50 Å / σ(F): 0 / % reflection Rfree: 5.2 % | ||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 14.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.308 / % reflection Rfree: 5.4 % / Rfactor Rwork: 0.283 |
