 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1klf | ||||||
|---|---|---|---|---|---|---|---|
| Title | FIMH ADHESIN-FIMC CHAPERONE COMPLEX WITH D-MANNOSE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | CHAPERONE/ADHESIN COMPLEX / ADHESIN-CHAPERONE COMPLEX / MANNOSE-BOUND / CHAPERONE-ADHESIN COMPLEX COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationpilus tip / mechanosensory behavior / cell adhesion involved in single-species biofilm formation / pilus / cell-substrate adhesion / D-mannose binding / host cell membrane / : / protein folding chaperone / cell wall organization ...pilus tip / mechanosensory behavior / cell adhesion involved in single-species biofilm formation / pilus / cell-substrate adhesion / D-mannose binding / host cell membrane / : / protein folding chaperone / cell wall organization / outer membrane-bounded periplasmic space / cell adhesion Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.79 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.79 Å | ||||||
 Authors Authors | Hung, C.S. / Bouckaert, J. | ||||||
 Citation Citation | Journal: Mol.Microbiol. / Year: 2002 Title: Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Authors: Hung, C.S. / Bouckaert, J. / Hung, D. / Pinkner, J. / Widberg, C. / DeFusco, A. / Auguste, C.G. / Strouse, R. / Langermann, S. / Waksman, G. / Hultgren, S.J. | ||||||
| History |
| ||||||
| Remark 300 | BIOMOLECULE: 1, 2, 3, 4, 5, 6, 7, 8 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH ...BIOMOLECULE: 1, 2, 3, 4, 5, 6, 7, 8 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 16CHAIN(S). SEE REMARK 350 FOR INFORMATION ON GENERATING THE BIOLOGICAL MOLECULE(S). THE BIOMOLECULES ARE 1, 2, 3, 4, 5, 6, 7 AND 8 RESPECTIVELY, EACH CONSISTING OF A COMPLEX OF CHAPERONE FIMC WITH ADHESIN FIMH. THERE IS NO BIOLOGICALLY SIGNIFICANT HIGHER OLIGOMERIZATION STATE. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1klf.cif.gz | 722.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1klf.ent.gz | 598.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1klf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kl/1klfftp://data.pdbj.org/pub/pdb/validation_reports/kl/1klf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1kiuC  1qunS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| 5 | 
| ||||||||
| 6 | 
| ||||||||
| 7 | 
| ||||||||
| 8 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | 1 chaperone - 1 adhesin - 1 mannose is one biological assembly |
-Components
| #1: Protein | Mass: 22724.049 Da / Num. of mol.: 8 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Protein | Mass: 29081.314 Da / Num. of mol.: 8 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #3: Sugar | ChemComp-MAN / 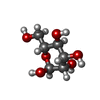  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 8 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 8Source method: isolated from a genetically manipulated source Formula: C6H12O6 #4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 495 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 495 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.48 Å3/Da / Density % sol: 40 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 8.2 Details: ammonium suplhate, pH 8.20, VAPOR DIFFUSION, SITTING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6.5 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-BM / Wavelength: 0.9793 / Wavelength: 0.9793 Å / Beamline: 19-BM / Wavelength: 0.9793 / Wavelength: 0.9793 Å |
| Detector | Type: CUSTOM-MADE / Detector: CCD / Date: Mar 18, 2001 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9793 Å / Relative weight: 1 |
| Reflection | Resolution: 2.79→43.68 Å / Num. all: 100438 / Num. obs: 99135 / % possible obs: 99.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.7 % / Biso Wilson estimate: 66.3 Å2 / Rmerge(I) obs: 0.069 / Rsym value: 0.069 / Net I/σ(I): 13 |
| Reflection shell | Resolution: 2.79→2.9 Å / Redundancy: 3.7 % / Rmerge(I) obs: 0.478 / Mean I/σ(I) obs: 2.7 / Rsym value: 0.478 / % possible all: 99.9 |
| Reflection | *PLUS Highest resolution: 2.8 Å / Lowest resolution: 50 Å / Num. measured all: 370427 |
| Reflection shell | *PLUS Highest resolution: 2.8 Å / Lowest resolution: 2.9 Å / % possible obs: 99.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1QUN Resolution: 2.79→43.68 Å / Rfactor Rfree error: 0.003 / Data cutoff high absF: 1364705.86 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 96.4 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.79→43.68 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: RESTRAINED / Weight Biso : 8 / Weight position: 250 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.79→2.96 Å / Rfactor Rfree error: 0.01 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.8 Å / Lowest resolution: 50 Å / Rfactor Rfree: 0.279 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.42 / Rfactor Rwork: 0.35 / Rfactor obs: 0.35 |
