 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1k2l: STRUCTURAL CHARACTERIZATION OF BISINTERCALATION IN HIGHER-ORDER D... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1k2l | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURAL CHARACTERIZATION OF BISINTERCALATION IN HIGHER-ORDER DNA AT A JUNCTION-LIKE QUADRUPLEX | ||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / DNA Strand Exchange / quadruplex / bisintercalation | Function / homology | Chem-BDC / : / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å  Authors AuthorsTeixeira, S.C.M. / Thorpe, J.H. / Todd, A.K. / Powell, H.R. / Adams, A. / Wakelin, L.P.G. / Denny, W.A. / Cardin, C.J. |  CitationJournal: J.MOL.BIOL. / Year: 2002 CitationJournal: J.MOL.BIOL. / Year: 2002Title: Structural Characterisation of Bisintercalation in Higher-order DNA at a Junction-like Quadruplex Authors: Teixeira, S.C.M. / Thorpe, J.H. / Todd, A.K. / Powell, H.R. / Adams, A. / Wakelin, L.P.G. / Denny, W.A. / Cardin, C.J. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1k2l.cif.gz | 19.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1k2l.ent.gz | 10.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1k2l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/k2/1k2lftp://data.pdbj.org/pub/pdb/validation_reports/k2/1k2l | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |
|




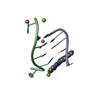

-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 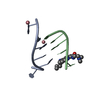
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 1809.217 Da / Num. of mol.: 2 / Source method: obtained synthetically #2: Chemical |   Mass: 58.933 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Co Mass: 58.933 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Co#3: Chemical | ChemComp-BDC / | 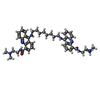  Mass: 726.952 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C44H54N8O2 Mass: 726.952 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C44H54N8O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 41.47 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 285 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: Sodium cacodylate buffer, magnesium acetate, cobalt chloride, spermine, MPD, pH 6.5, VAPOR DIFFUSION, SITTING DROP, temperature 285.0K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 285 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X31 / Wavelength: 0.979 Å / Beamline: X31 / Wavelength: 0.979 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Nov 15, 1996 |
| Radiation | Monochromator: Mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 2.28→13.5 Å / Num. all: 2063 / Num. obs: 2063 / % possible obs: 88.06 % / Observed criterion σ(F): 2 / Observed criterion σ(I): -3 / Rmerge(I) obs: 0.071 / Net I/σ(I): 11.57 |
| Reflection shell | Resolution: 2.28→2.36 Å / % possible all: 93.8 |
| Reflection | *PLUS Highest resolution: 2.35 Å / Rmerge(I) obs: 0.074 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.4→9 Å / Num. parameters: 848 / Num. restraintsaints: 1140 / Cross valid method: FREE R / σ(F): 4 / Stereochemistry target values: ENGH AND HUBER
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 / Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 264 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→9 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.48 Å /
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / Lowest resolution: 9 Å / % reflection Rfree: 5 % / Rfactor Rfree: 0.3094 / Rfactor Rwork: 0.2385 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: s_angle_d |
