 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jro: Crystal Structure of Xanthine Dehydrogenase from Rhodobacter caps... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jro | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Xanthine Dehydrogenase from Rhodobacter capsulatus | ||||||
 Components Components | (xanthine dehydrogenase, chain ...) x 2 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / Partial Beta-Barrel / xdh / xo | ||||||
| Function / homology |  Function and homology information Function and homology information1.1.1.204 / xanthine dehydrogenase activity / molybdenum ion binding / FAD binding / 2 iron, 2 sulfur cluster binding / oxidoreductase activity / iron ion binding Similarity search - Function | ||||||
| Biological species |  Rhodobacter capsulatus (bacteria) Rhodobacter capsulatus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Truglio, J.J. / Theis, K. / Leimkuhler, S. / Rappa, R. / Rajagopalan, K.V. / Kisker, C. | ||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: Crystal structures of the active and alloxanthine-inhibited forms of xanthine dehydrogenase from Rhodobacter capsulatus Authors: Truglio, J.J. / Theis, K. / Leimkuhler, S. / Rappa, R. / Rajagopalan, K.V. / Kisker, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jro.cif.gz | 911 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jro.ent.gz | 735.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1jro.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jr/1jroftp://data.pdbj.org/pub/pdb/validation_reports/jr/1jro | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1jrpC  1fo4S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |

-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| 2 | 
| ||||||||||
| Unit cell |
|
-Components
-Xanthine dehydrogenase, chain ... , 2 types, 8 molecules ACEGBDFH
| #1: Protein | Mass: 49347.453 Da / Num. of mol.: 4 / Fragment: chain A, residues 1-462 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Rhodobacter capsulatus (bacteria) / Production host: #2: Protein | Mass: 82978.031 Da / Num. of mol.: 4 / Fragment: chain B, residues 1-777 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Rhodobacter capsulatus (bacteria) / Production host: |
|---|
-Non-polymers , 6 types, 147 molecules 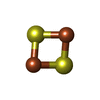


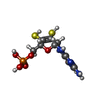


| #3: Chemical | ChemComp-FES /  Mass: 175.820 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Fe2S2 Mass: 175.820 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Fe2S2#4: Chemical | ChemComp-FAD /  Mass: 785.550 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM#5: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#6: Chemical | ChemComp-MTE /  Mass: 395.352 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H14N5O6PS2 Mass: 395.352 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H14N5O6PS2#7: Chemical | ChemComp-MOS /  Mass: 161.012 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: HMoO2S Mass: 161.012 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: HMoO2S#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 123 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.2 Å3/Da / Density % sol: 44 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 8 Details: PEG, Tris, DTT, isopropanol at pH 8.0, VAPOR DIFFUSION, HANGING DROP at 295K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| |||||||||||||||
| Detector |
| |||||||||||||||
| Radiation |
| |||||||||||||||
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 | |||||||||||||||
| Reflection | Resolution: 2.7→50 Å / Num. all: 187987 / Num. obs: 187987 / % possible obs: 99.2 % / Observed criterion σ(I): -1000 / Redundancy: 3.8 % / Rsym value: 0.17 / Net I/σ(I): 11.2 | |||||||||||||||
| Reflection shell | Resolution: 2.7→2.8 Å / % possible obs: 98 % / Mean I/σ(I) obs: 2.7 / Rsym value: 0.73 | |||||||||||||||
| Reflection | *PLUS Num. measured all: 722868 / Rmerge(I) obs: 0.161 | |||||||||||||||
| Reflection shell | *PLUS Rmerge(I) obs: 0.73 |

- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1FO4 Resolution: 2.7→50 Å / Cor.coef. Fo:Fc: 0.92 / Cor.coef. Fo:Fc free: 0.887 / SU B: 11.3 / SU ML: 0.2 / SU Rfree: 0.3 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.6 Details: FOUR-FOLD NCS RESTRAINTS WERE IMPOSED ON CHAINS A, B, C, D AND E, F, G, H
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: BABINET MODEL WITH MAS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 39.67 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.7→2.77 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Version: 5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 39.67 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.377 / Rfactor Rwork: 0.286 |
