 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jot: STRUCTURE OF THE LECTIN MPA COMPLEXED WITH T-ANTIGEN DISACCHARIDE -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jot | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF THE LECTIN MPA COMPLEXED WITH T-ANTIGEN DISACCHARIDE | |||||||||
 Components Components | (AGGLUTININ) x 2 | |||||||||
 Keywords Keywords | LECTIN / MULTI-WAVELENGTH ANOMALOUS DIFFRACTION (MAD) / T-ANTIGEN / MACLURA POMIFERA / BETA PRISM | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  Maclura pomifera (Osage orange) Maclura pomifera (Osage orange) | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | |||||||||
 Authors Authors | Lee, X. / Thompson, A. / Zhang, Z. / Hoa, T.-T. / Biesterfeldt, J. / Ogata, C. / Xu, L. / Johnston, R.A.Z. / Young, N.M. | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1998 Title: Structure of the complex of Maclura pomifera agglutinin and the T-antigen disaccharide, Galbeta1,3GalNAc. Authors: Lee, X. / Thompson, A. / Zhang, Z. / Ton-that, H. / Biesterfeldt, J. / Ogata, C. / Xu, L. / Johnston, R.A. / Young, N.M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jot.cif.gz | 45.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jot.ent.gz | 30.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1jot.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1jot_validation.pdf.gz | 801.9 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1jot_full_validation.pdf.gz | 804.1 KB | Display | |
| Data in XML | 1jot_validation.xml.gz | 9.3 KB | Display | |
| Data in CIF | 1jot_validation.cif.gz | 12.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jo/1jotftp://data.pdbj.org/pub/pdb/validation_reports/jo/1jot | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 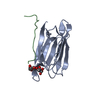
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 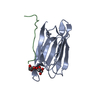
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14768.595 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: FROM THE SEEDS OF THE MORACEAE PLANT FAMILY / Source: (natural) Maclura pomifera (Osage orange) / Organ: SEEDS / References: UniProt: P18674 |
|---|---|
| #2: Protein/peptide | Mass: 2144.373 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: FROM THE SEEDS OF THE MORACEAE PLANT FAMILY / Source: (natural) Maclura pomifera (Osage orange) / Organ: SEEDS / References: UniProt: P18676 |
| #3: Polysaccharide | beta-D-galactopyranose-(1-3)-2-acetamido-2-deoxy-alpha-D-galactopyranose / Thomsen-Friedenreich antigen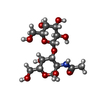  Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: Thomsen-Friedenreich antigen |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 91 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 91 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.83 Å3/Da / Density % sol: 56 % |
|---|---|
| Crystal grow | Method: vapor diffusion, sitting drop / pH: 4.5 Details: CONVENTIONAL VAPOR DIFFUSION OF SITTING DROP. 1.0M OF LITHIUM SULFATE BUFFERED WITH 0.2 SODIUM CITRATE AT PH4.5 IN RESERVOIR DROPS MADE OF 15 MICROLITERS OF PROTEIN SOLUTION (10.0 MG/ML) AND ...Details: CONVENTIONAL VAPOR DIFFUSION OF SITTING DROP. 1.0M OF LITHIUM SULFATE BUFFERED WITH 0.2 SODIUM CITRATE AT PH4.5 IN RESERVOIR DROPS MADE OF 15 MICROLITERS OF PROTEIN SOLUTION (10.0 MG/ML) AND EQUAL VOLUMN OF RESERVOIR SOLUTION. ROOM TEMPERATURE., vapor diffusion - sitting drop |
| Crystal grow | *PLUS Method: unknown |
-Data collection
| Diffraction | Mean temperature: 300 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5434 |
| Detector | Type: XUONG-HAMLIN MULTIWIRE / Detector: AREA DETECTOR / Date: Jul 1, 1991 |
| Radiation | Monochromator: YES / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5434 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→30 Å / Num. obs: 10135 / % possible obs: 93.9999 % / Observed criterion σ(I): 2 / Redundancy: 9.6 % / Rmerge(I) obs: 0.036 / Net I/σ(I): 7.84 |
| Reflection shell | Resolution: 2.2→3.99 Å |
| Reflection | *PLUS % possible obs: 93.5 % / Num. measured all: 98232 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: STRUCTURE OBTAINED FROM MAD Resolution: 2.2→6 Å / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.3 Å / Total num. of bins used: 6 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.854 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.236 |
