 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jlx | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | AGGLUTININ IN COMPLEX WITH T-DISACCHARIDE | |||||||||
 Components Components | AGGLUTININ | |||||||||
 Keywords Keywords | LECTIN / COMPLEX (LECTIN-SACCHARIDE) / T-DISACCHARIDE HOMODIMER / BIVALENT | |||||||||
| Function / homology |  Function and homology information Function and homology informationAgglutinin domain / Agglutinin domain superfamily / Agglutinin domain / Agglutinin / : / Trefoil (Acidic Fibroblast Growth Factor, subunit A) - #50 / Trefoil (Acidic Fibroblast Growth Factor, subunit A) / Trefoil / Mainly Beta Similarity search - Domain/homology | |||||||||
| Biological species |  Amaranthus caudatus (amaranth) Amaranthus caudatus (amaranth) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | |||||||||
 Authors Authors | Transue, T.R. / Smith, A.K. / Mo, H. / Goldstein, I.J. / Saper, M.A. | |||||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1997 Title: Structure of benzyl T-antigen disaccharide bound to Amaranthus caudatus agglutinin. Authors: Transue, T.R. / Smith, A.K. / Mo, H. / Goldstein, I.J. / Saper, M.A. #1: Journal: J.Biol.Chem. / Year: 1989Title: Isolation and Characterization of Amaranthin, a Lectin Present in the Seeds of Amaranthus Caudatus, that Recognizes the T-(or Cryptic T)-Antigen Authors: Rinderle, S.J. / Goldstein, I.J. / Matta, K.L. / Ratcliffe, R.M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jlx.cif.gz | 135.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jlx.ent.gz | 109.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1jlx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jl/1jlxftp://data.pdbj.org/pub/pdb/validation_reports/jl/1jlx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1jlySC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 34812.324 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Amaranthus caudatus (amaranth) / Organ: SEED / References: PIR: S24263, UniProt: Q71QF2*PLUS#2: Polysaccharide | 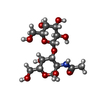  Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: Thomsen-Friedenreich antigen #3: Chemical | ChemComp-FOR /   Mass: 30.026 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH2O Mass: 30.026 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH2O#4: Chemical | 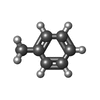  Mass: 92.138 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H8 Mass: 92.138 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H8#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 297 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 297 / Source method: isolated from a natural source / Formula: H2OSequence details | THE SEQUENCE WAS DEDUCED FROM THE KNOWN SEQUENCE OF AN APPARENT HOMOLOGUE FROM AMARANTHUS ...THE SEQUENCE WAS DEDUCED FROM THE KNOWN SEQUENCE OF AN APPARENT HOMOLOGUE FROM AMARANTHUS | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 4 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.66 Å3/Da / Density % sol: 57 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.7 / Details: pH 6.7 | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 273 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: A1 / Wavelength: 0.908 / Beamline: A1 / Wavelength: 0.908 |
| Detector | Detector: CCD / Date: Aug 7, 1994 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.908 Å / Relative weight: 1 |
| Reflection | Num. obs: 33071 / % possible obs: 88.4 % / Observed criterion σ(I): 0 / Redundancy: 4.1 % / Rmerge(I) obs: 0.109 / Net I/σ(I): 11.3 |
| Reflection shell | Highest resolution: 2.2 Å / Rmerge(I) obs: 0.272 / Mean I/σ(I) obs: 7.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1JLY Resolution: 2.2→10 Å / σ(F): 0 Details: THE N-TERMINAL ALA 1 APPEARS TO HAVE DENSITY CONSISTENT WITH A FORMYL GROUP ATTACHED TO THE N-TERMINUS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.06 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
