 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jkx: Unexpected formation of an epoxide-derived multisubstrate adduct ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jkx | ||||||
|---|---|---|---|---|---|---|---|
| Title | Unexpected formation of an epoxide-derived multisubstrate adduct inhibitor on the active site of GAR transformylase | ||||||
 Components Components | PHOSPHORIBOSYLGLYCINAMIDE FORMYLTRANSFERASE | ||||||
 Keywords Keywords | TRANSFERASE / PURINE BIOSYNTHESIS / ANTI-CANCER AGENT / ENZYME-ASSEMBLED MULTISUBSTRATE ADDUCT INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationphosphoribosylglycinamide formyltransferase 1 / phosphoribosylglycinamide formyltransferase activity / 'de novo' IMP biosynthetic process / DNA damage response / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Greasley, S.E. / Marsilje, T.H. / Cai, H. / Baker, S. / Benkovic, S.J. / Boger, D.L. / Wilson, I.A. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2001 Title: Unexpected formation of an epoxide-derived multisubstrate adduct inhibitor on the active site of GAR transformylase. Authors: Greasley, S.E. / Marsilje, T.H. / Cai, H. / Baker, S. / Benkovic, S.J. / Boger, D.L. / Wilson, I.A. #1: Journal: Bioorg.Med.Chem. / Year: 1997Title: Functionalized analogues of 5,8,10-trideazafolate: Development of an enzyme-assembled tight binding inhibitor of GAR Tfase and a potential irreversible inhibitor of AICAR Tfase Authors: Boger, D.L. / Haynes, N.-E. / Warren, M.S. / Ramcharan, J. / Kitos, P.A. / Benkovic, S.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jkx.cif.gz | 186.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jkx.ent.gz | 149.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1jkx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jk/1jkxftp://data.pdbj.org/pub/pdb/validation_reports/jk/1jkx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1c2tS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| 2 | 
| ||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23266.254 Da / Num. of mol.: 4 / Fragment: TRANSFERASE Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P08179, phosphoribosylglycinamide formyltransferase 1 #2: Chemical | ChemComp-138 / 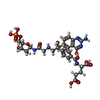  Mass: 752.620 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C30H37N6O15P Mass: 752.620 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C30H37N6O15PDetails: MAI derived from beta-GAR and the epoxide derivative of 10-bromo-10-bromomethyl-5,8,10-trideazafolic acid #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 607 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 607 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.36 Å3/Da / Density % sol: 47.93 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 7.4 Details: PEG 3350, CaCl2, MPD, imidazole malate, PH 7.4, VAPOR DIFFUSION, SITTING DROP at 295K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / Details: used macroseeding | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 95 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.98 Å / Beamline: BL9-1 / Wavelength: 0.98 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Apr 12, 1998 / Details: MIRRORS |
| Radiation | Monochromator: Flat mirror (vertical focusing); single crystal Si(311) bent monochromator (horizontal focusing) Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→30 Å / Num. obs: 106828 / % possible obs: 94.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 1.9 % / Biso Wilson estimate: 22.1 Å2 / Rmerge(I) obs: 0.05 / Net I/σ(I): 6.2 |
| Reflection shell | Resolution: 1.6→1.64 Å / Redundancy: 1.5 % / Rmerge(I) obs: 0.325 / Mean I/σ(I) obs: 2.2 / Num. unique all: 6398 / % possible all: 76.4 |
| Reflection | *PLUS Rmerge(I) obs: 0.05 |
| Reflection shell | *PLUS % possible obs: 76.4 % / Num. unique obs: 6398 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PBD ENTRY 1C2T Resolution: 1.6→30 Å / Rfactor Rfree error: 0.002 / Data cutoff high absF: 809232.51 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 38.1431 Å2 / ksol: 0.355482 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.8 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→30 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.7 Å / Rfactor Rfree error: 0.008 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 10 % | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 23.8 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.32 / % reflection Rfree: 10.1 % / Rfactor Rwork: 0.299 |
