 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jhg | ||||||
|---|---|---|---|---|---|---|---|
| Title | TRP REPRESSOR MUTANT V58I | ||||||
 Components Components | TRP OPERON REPRESSOR | ||||||
 Keywords Keywords | DNA BINDING PROTEIN / REGULATORY PROTEIN-PEPTIDE complex | ||||||
| Function / homology |  Function and homology information Function and homology informationsequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / regulation of DNA-templated transcription / DNA-templated transcription / DNA binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / ISOMORPHOUS TO PDB ENTRY 2WRP / Resolution: 1.3 Å X-RAY DIFFRACTION / SYNCHROTRON / ISOMORPHOUS TO PDB ENTRY 2WRP / Resolution: 1.3 Å | ||||||
 Authors Authors | Lawson, C.L. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1996 Title: An atomic view of the L-tryptophan binding site of trp repressor. Authors: Lawson, C.L. #1: Journal: Biological Structure and Dynamics: Proceedings of the Ninth Conversation in the Discipline Biomolecular Stereodynamics, Held at the State University of New York at Albany, June 20-24, 1995 Year: 1996 Title: Structural Consequences of Twp Methyl Additions in the Escherichia Coli Trp Repressor L-Tryptophan Binding Pocket Authors: Lawson, C.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jhg.cif.gz | 58.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jhg.ent.gz | 43.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1jhg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jh/1jhgftp://data.pdbj.org/pub/pdb/validation_reports/jh/1jhg | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 11578.260 Da / Num. of mol.: 1 / Mutation: CHAIN A, V58I Source method: isolated from a genetically manipulated source Source: (gene. exp.) Description: I58 TRP REPRESSOR WAS GROWN IN ESCHERICHIA COLI STRAIN BL21 (DE3) USING T7 EXPRESSION SYSTEM VECTORS; Cell line: BL21 / Plasmid: PET13A / Species (production host): Escherichia coli / Cell (production host): CYTOPLASM / Gene (production host): TRPRI58 / Production host: |
|---|---|
| #2: Chemical | ChemComp-PO4 /   Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 |
| #3: Chemical | ChemComp-TRP / 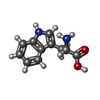  Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12N2O2 Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12N2O2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 119 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 119 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | TWO TORSIONAL ANGLES IN THE L-TRYPTOPHAN BINDING POCKET HAVE DISTINCTLY NON-IDEAL GEOMETRY: CHI1 OF ...TWO TORSIONAL ANGLES IN THE L-TRYPTOPHAN |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.9 Å3/Da / Density % sol: 37 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 5 Details: HANGING DROP VAPOR DIFFUSION 2.0 M SODIUM PHOSPHATE, PH 5.0 600 MM AMMONIUM CHLORIDE 5 MM L-TRYPTOPHAN, vapor diffusion - hanging drop | ||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 287 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 0.99 / Beamline: X12C / Wavelength: 0.99 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE AREA DETECTOR / Date: Aug 1, 1994 |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.99 Å / Relative weight: 1 |
| Reflection | Resolution: 1.3→30 Å / Num. obs: 86193 / % possible obs: 97.4 % / Observed criterion σ(I): 0.25 / Redundancy: 3.6 % / Rmerge(I) obs: 0.032 / Net I/σ(I): 28.4 |
| Reflection shell | Resolution: 1.3→1.35 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.167 / Mean I/σ(I) obs: 20 / % possible all: 94 |
| Reflection shell | *PLUS % possible obs: 94 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: ISOMORPHOUS TO PDB ENTRY 2WRP Resolution: 1.3→30 Å / Num. parameters: 8337 / Num. restraintsaints: 2568 / Cross valid method: FREE R / Stereochemistry target values: ENGH AND HUBER Details: RIDING HYDROGEN ATOM OPTION OF SHELXL USED, ANISOTROPIC TEMPERATURE FACTORS REFINED BUT NOT INCLUDED IN THE DEPOSITION. SOME SIDE-CHAIN, SOLVENT, AND ALL PHOSPHATE ATOMS HAVE PARTIAL ...Details: RIDING HYDROGEN ATOM OPTION OF SHELXL USED, ANISOTROPIC TEMPERATURE FACTORS REFINED BUT NOT INCLUDED IN THE DEPOSITION. SOME SIDE-CHAIN, SOLVENT, AND ALL PHOSPHATE ATOMS HAVE PARTIAL OCCUPANCIES. APPARENT "SHORT" INTERATOMIC DISTANCES INDICATE POSITIONS IN THE CRYSTAL WHERE THERE ARE AT LEAST TWO ALTERNATE MODELS.
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: SWAT 7.731 2.0 (BABINET'S PRINCIPLE) | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 15 / Occupancy sum hydrogen: 4248 / Occupancy sum non hydrogen: 3504 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.3→30 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
