 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jdt: CRYSTAL STRUCTURE OF 5'-DEOXY-5'-METHYLTHIOADENOSINE PHOSPHORYLAS... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jdt | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF 5'-DEOXY-5'-METHYLTHIOADENOSINE PHOSPHORYLASE COMPLEXED WITH MTA AND SULFATE ION | ||||||
 Components Components | 5'-METHYLTHIOADENOSINE PHOSPHORYLASE | ||||||
 Keywords Keywords | TRANSFERASE / alpha-beta protein | ||||||
| Function / homology |  Function and homology information Function and homology informationS-methyl-5'-thioadenosine phosphorylase / S-methyl-5-thioadenosine phosphorylase activity / nucleoside catabolic process / purine-nucleoside phosphorylase / cytosol Similarity search - Function | ||||||
| Biological species |   Sulfolobus solfataricus (archaea) Sulfolobus solfataricus (archaea) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Appleby, T.C. / Mathews, I.I. / Porcelli, M. / Cacciapuoti, G. / Ealick, S.E. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2001 Title: Three-dimensional structure of a hyperthermophilic 5'-deoxy-5'-methylthioadenosine phosphorylase from Sulfolobus solfataricus. Authors: Appleby, T.C. / Mathews, I.I. / Porcelli, M. / Cacciapuoti, G. / Ealick, S.E. #1: Journal: J.Biol.Chem. / Year: 1994Title: Purification and characterization of extremely thermophilic and thermostable 5'-methylthioadenosine phosphorylase from the archaeon Sulfolobus solfataricus. Purine nucleoside phosphorylase ...Title: Purification and characterization of extremely thermophilic and thermostable 5'-methylthioadenosine phosphorylase from the archaeon Sulfolobus solfataricus. Purine nucleoside phosphorylase activity and evidence for intersubunit disulfide bonds Authors: Cacciapuoti, G. / Porcelli, M. / Bertoldo, C. / De Rosa, M. / Zappia, V. #2: Journal: J.Biol.Chem. / Year: 1990Title: Three-Dimensional Structure of Human Erythrocytic Purine Nucleoside Phosphorylase At 3.2 Resolution Authors: Ealick, S.E. / Rule, S.A. / Carter, D.C. / Greenhough, T.J. / Babu, Y.S. #3: Journal: Structure / Year: 1999Title: The structure of human 5'-deoxy-5'-methylthioadenosine phosphorylase at 1.7 resolution provides insights into substrate binding and catalysis Authors: Appleby, T.C. / Erion, M.D. / Ealick, S.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jdt.cif.gz | 145.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jdt.ent.gz | 115.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1jdt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jd/1jdtftp://data.pdbj.org/pub/pdb/validation_reports/jd/1jdt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1jdsC  1jduC  1jdvC  1jdzC  1je0C  1je1C  1jp7C  1jpvC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| Unit cell |
| ||||||||||
| Details | hexamer generated from the trimer in the asymmetric unit by the crystallographic two fold |
-Components
| #1: Protein | Mass: 25763.457 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Sulfolobus solfataricus (archaea) / Production host:  References: UniProt: P50389, S-methyl-5'-thioadenosine phosphorylase #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#3: Chemical | 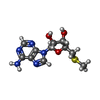  Mass: 297.334 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H15N5O3S Mass: 297.334 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H15N5O3S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 167 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 167 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.52 Å3/Da / Density % sol: 51.24 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.4 Details: dioxane, MPD, MgCl2, NaCl, Tris.HCl at pH 7.4, VAPOR DIFFUSION, HANGING DROP, 291K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Details: used microseeding | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: A1 / Wavelength: 0.9023 Å / Beamline: A1 / Wavelength: 0.9023 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9023 Å / Relative weight: 1 |
| Reflection | Resolution: 2→30 Å / Num. all: 50489 / Num. obs: 50489 / % possible obs: 95.2 % / Observed criterion σ(I): 1 / Redundancy: 4.2 % / Rmerge(I) obs: 0.074 / Net I/σ(I): 7.1 |
| Reflection shell | Highest resolution: 2 Å / Rmerge(I) obs: 0.487 / Mean I/σ(I) obs: 1.5 / % possible all: 79.2 |
| Reflection | *PLUS Num. measured all: 213837 |
| Reflection shell | *PLUS % possible obs: 72 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: native SsMTAP Resolution: 2→20 Å / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 2 / σ(I): 1 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.7 Å2 | |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→20 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 20 Å / σ(F): 2 / % reflection Rfree: 5 % / Rfactor obs: 0.222 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 34.7 Å2 |
