 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1i1w: 0.89A Ultra high resolution structure of a Thermostable Xylanase ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1i1w | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | 0.89A Ultra high resolution structure of a Thermostable Xylanase from Thermoascus Aurantiacus | |||||||||
 Components Components | ENDO-1,4-BETA-XYLANASE | |||||||||
 Keywords Keywords | HYDROLASE / XYLAN DEGRADATION / GLYCOSIDASE / ENZYME / ULTRA HIGH RESOLUTION / CRYO TEMPERATURE / 1 / 4-BETA-XYLAN XYLANOHYDROLASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationendo-1,4-beta-xylanase / endo-1,4-beta-xylanase activity / xylan catabolic process Similarity search - Function | |||||||||
| Biological species |  Thermoascus aurantiacus (fungus) Thermoascus aurantiacus (fungus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.89 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.89 Å | |||||||||
 Authors Authors | Natesh, R. / Ramakumar, S. / Viswamitra, M.A. | |||||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2003 Title: Thermostable xylanase from Thermoascus aurantiacus at ultrahigh resolution (0.89 A) at 100 K and atomic resolution (1.11 A) at 293 K refined anisotropically to small-molecule accuracy. Authors: Natesh, R. / Manikandan, K. / Bhanumoorthy, P. / Viswamitra, M.A. / Ramakumar, S. #1: Journal: J.Mol.Biol. / Year: 1999Title: Crystal Structure at 1.8 A Resolution and Proposed Amino Acid Sequence of a Thermostable Xylanase from Thermoascus Aurantiascus Authors: Natesh, R. / Bhanumoorthy, P. / Vithayathil, P.J. / Sekar, K. / Ramakumar, S. / Viswamitra, M.A. #2: Journal: J.Mol.Biol. / Year: 1993Title: Crystallization and Preliminary X-Ray Diffraction Analysis of Crystals of Thermoascus Aurantiacus Xylanase Authors: Viswamitra, M.A. / Bhanumoorthy, P. / Ramakumar, S. / Manjula, M.V. / Vithayathil, P.J. / Murthy, S.K. / Naren, A.P. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1i1w.cif.gz | 193.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1i1w.ent.gz | 153.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1i1w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i1/1i1wftp://data.pdbj.org/pub/pdb/validation_reports/i1/1i1w | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 32868.734 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Thermoascus aurantiacus (fungus) / Strain: STRAIN ISOLATED FROM LOCAL INDIAN SOIL / References: UniProt: P23360, endo-1,4-beta-xylanase |
|---|
-Non-polymers , 5 types, 436 molecules 

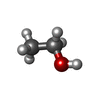
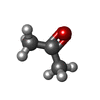

| #2: Chemical | ChemComp-UNX /  Num. of mol.: 13 / Source method: obtained synthetically Num. of mol.: 13 / Source method: obtained synthetically#3: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical |  Mass: 46.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O Mass: 46.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O#5: Chemical | ChemComp-ACN / |  Mass: 58.079 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H6O Mass: 58.079 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H6O#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 414 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.95 Å3/Da / Density % sol: 36.81 % Description: STARTING MODEL WAS LOCALLY AVAILABLE INTERMEDIATE REFINED 1.11 A ROOM TEMPERATURE MODEL. THE STRANGE HETERO MOLECULES SEEN IN 0.89 A HIGH RESOLUTION MAPS, labelled as "UNX 1406 TO UNX ...Description: STARTING MODEL WAS LOCALLY AVAILABLE INTERMEDIATE REFINED 1.11 A ROOM TEMPERATURE MODEL. THE STRANGE HETERO MOLECULES SEEN IN 0.89 A HIGH RESOLUTION MAPS, labelled as "UNX 1406 TO UNX 1410" and "UNX 1411 TO UNX 1418", ARE YET TO BE IDENTIFIED. EOH AND ACN WERE ASSIGNED BASED ON UNRESTRAINED BOND LENGTH AND ANGLES. | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7.2 Details: Ammonium Sulphate, TRIS/HCl, pH 7.2, VAPOR DIFFUSION, SITTING DROP, temperature 293.0K | ||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / Details: Viswamitra, M.A., (1993) J.Mol.Biol., 232, 987. | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X9B / Wavelength: 0.98 / Wavelength: 0.98 Å / Beamline: X9B / Wavelength: 0.98 / Wavelength: 0.98 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Apr 3, 1999 / Details: MIRROR |
| Radiation | Monochromator: DOUBLE CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 0.89→35 Å / Num. obs: 177476 / % possible obs: 92 % / Redundancy: 4.8 % / Rmerge(I) obs: 0.047 / Net I/σ(I): 32.65 |
| Reflection shell | Resolution: 0.89→0.92 Å / Rmerge(I) obs: 0.18 / Mean I/σ(I) obs: 7.82 / % possible all: 83 |
| Reflection | *PLUS Lowest resolution: 35 Å / % possible obs: 92 % / Redundancy: 4.8 % / Num. measured all: 858643 |
| Reflection shell | *PLUS % possible obs: 83 % / Rmerge(I) obs: 0.18 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: LOCALLY AVAILABLE INTERMEDIATE REFINED ROOM TEMP. 1.11 A MODEL Resolution: 0.89→10 Å / Num. parameters: 24867 / Num. restraintsaints: 1 / Cross valid method: FREE R / σ(F): 0 StereochEM target val spec case: FINAL 18 CYCLES OF BLOCKED L.S. REFINEMENT (2 CYCLES ON EACH OF 9 BLOCKS) WAS PERFORMED USING SHELXL WITHOUT ANY RESTRAINS. Stereochemistry target values: ENGH AND HUBER Details: AS THE MOLECULE WAS REFINED WITH NO RESTRAINTS THE NON PLANARITY OF THE ARG 124 B CONFORMER AMIDE GROUP MAY NOT BE TAKEN AS TRUE SINCE THE PLANARITY RESTRAINTS WERE NOT IMPOSED TO THIS ...Details: AS THE MOLECULE WAS REFINED WITH NO RESTRAINTS THE NON PLANARITY OF THE ARG 124 B CONFORMER AMIDE GROUP MAY NOT BE TAKEN AS TRUE SINCE THE PLANARITY RESTRAINTS WERE NOT IMPOSED TO THIS SPARCINGLY OCCUPIED DISORDERED ALTERNATE CONFORMER B. CNS 0.4 was also used for refinement.
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201- | |||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 51 / Occupancy sum hydrogen: 2125 / Occupancy sum non hydrogen: 2765.87 | |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.89→10 Å
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 0.89→0.92 Å | |||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 10 Å / Rfactor Rwork: 0.09 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
