 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hyt: RE-DETERMINATION AND REFINEMENT OF THE COMPLEX OF BENZYLSUCCINIC ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hyt | ||||||
|---|---|---|---|---|---|---|---|
| Title | RE-DETERMINATION AND REFINEMENT OF THE COMPLEX OF BENZYLSUCCINIC ACID WITH THERMOLYSIN AND ITS RELATION TO THE COMPLEX WITH CARBOXYPEPTIDASE A | ||||||
 Components Components | THERMOLYSIN | ||||||
 Keywords Keywords | HYDROLASE(METALLOPROTEINASE) | ||||||
| Function / homology |  Function and homology information Function and homology informationthermolysin / metalloendopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR SUBSTITUTION / Resolution: 1.7 Å X-RAY DIFFRACTION / MOLECULAR SUBSTITUTION / Resolution: 1.7 Å | ||||||
 Authors Authors | Hausrath, A.C. / Matthews, B.W. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1994 Title: Redetermination and refinement of the complex of benzylsuccinic acid with thermolysin and its relation to the complex with carboxypeptidase A. Authors: Hausrath, A.C. / Matthews, B.W. #1: Journal: Biochemistry / Year: 1984Title: Binding of N-Carboxymethyl Dipeptide Inhibitors to Thermolysin Determined by X-Ray Crystallography: A Novel Class of Transition-State Analogues for Zinc Peptidases Authors: Monzingo, A.F. / Matthews, B.W. #2: Journal: J.Mol.Biol. / Year: 1982Title: Structure of Thermolysin Refined at 1.6 Angstroms Resolution Authors: Holmes, M.A. / Matthews, B.W. #3: Journal: J.Biol.Chem. / Year: 1974Title: The Conformation of Thermolysin Authors: Matthews, B.W. / Weaver, L.H. / Kester, W.R. #4: Journal: Nature New Biol. / Year: 1972Title: Structure of Thermolysin Authors: Matthews, B.W. / Colman, P.M. / Jansonius, J.N. / Titani, K. / Walsh, K.A. / Neurath, H. #5: Journal: Nature New Biol. / Year: 1972Title: Three Dimensional Structure of Thermolysin Authors: Matthews, B.W. / Jansonius, J.N. / Colman, P.M. / Schoenborn, B.P. / Duporque, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hyt.cif.gz | 79.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hyt.ent.gz | 58.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1hyt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hy/1hytftp://data.pdbj.org/pub/pdb/validation_reports/hy/1hyt | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|




















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: CIS PROLINE - PRO 51 |
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 34362.305 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P00800, thermolysin |
|---|
-Non-polymers , 5 types, 164 molecules 


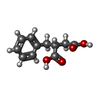

| #2: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-ZN / |  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-DMS / |  Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#5: Chemical | ChemComp-BZS / |  Mass: 208.211 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12O4 Mass: 208.211 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12O4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.42 Å3/Da / Density % sol: 49.25 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.2 / Details: pH 7.2 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown / Details: Holmes, M.A., (1982) J.Mol.Biol., 160, 623. | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: XUONG-HAMLIN / Detector: MULTIWIRE AREA DETECTOR |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE CRYSTAL / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.7 Å / Num. obs: 35931 / % possible obs: 94 % / Rmerge(I) obs: 0.045 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR SUBSTITUTION / Resolution: 1.7→20 Å / σ(F): 4 /
| ||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→20 Å
| ||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Classification: refinement | ||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.157 | ||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 30.1 Å2 | ||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: t_angle_d / Dev ideal: 1.9 |
