 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hn2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF BOVINE OBP COMPLEXED WITH AMINOANTHRACENE | ||||||
 Components Components | ODORANT-BINDING PROTEIN | ||||||
 Keywords Keywords | PROTEIN BINDING / olfaction / odorant binding protein / aminoanthracene | ||||||
| Function / homology |  Function and homology information Function and homology informationodorant binding / small molecule binding / sensory perception of smell / : Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / molecular replacement / Resolution: 1.8 Å X-RAY DIFFRACTION / molecular replacement / Resolution: 1.8 Å | ||||||
 Authors Authors | Vincent, F. / Spinelli, S. / Tegoni, M. / Cambillau, C. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2001 Title: The insect attractant 1-octen-3-ol is the natural ligand of bovine odorant-binding protein. Authors: Ramoni, R. / Vincent, F. / Grolli, S. / Conti, V. / Malosse, C. / Boyer, F.D. / Nagnan-Le Meillour, P. / Spinelli, S. / Cambillau, C. / Tegoni, M. | ||||||
| History |
| ||||||
| Remark 600 | heterogen The Native Bovine OBP has a natural ligand Octene-3-ol (PDB ID 1G85). In this experiment, ...heterogen The Native Bovine OBP has a natural ligand Octene-3-ol (PDB ID 1G85). In this experiment, the native Bovine OBP was soaked with 2mM Aminoanthracene, which removed all of the natural ligand in cavity A (giving all Aminoantracene atoms occupancy 1.0) but left 40% of the natural ligand in cavity B. The occupancy of aminoanthracene atoms in cavity B was 0.60 because of the presence of octene-3-ol, with the R and S enantiomers each having occupancies of 0.20. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hn2.cif.gz | 81.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hn2.ent.gz | 60.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1hn2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hn/1hn2ftp://data.pdbj.org/pub/pdb/validation_reports/hn/1hn2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1g85C  1obpS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18528.381 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) #2: Chemical | 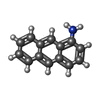  Mass: 193.244 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H11N Mass: 193.244 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H11N#3: Chemical | ChemComp-3OL / ( |   Mass: 128.212 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H16O Mass: 128.212 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H16O#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 120 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 120 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.04 Å3/Da / Density % sol: 39.72 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.7 Details: dialysys against 38% ethanol in 25 mM sodium Citrate at 4 degrees, pH 5.7 | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 5.4 / Method: microdialysis | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 28, 2000 |
| Radiation | Monochromator: Osmic mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→17 Å / Num. obs: 26203 / % possible obs: 92 % / Redundancy: 2.8 % / Biso Wilson estimate: 21.4 Å2 / Rsym value: 5.2 / Net I/σ(I): 8.5 |
| Reflection shell | Resolution: 1.8→1.89 Å / Redundancy: 2.8 % / Mean I/σ(I) obs: 2.1 / Rsym value: 0.44 / % possible all: 85.4 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 18 Å / % possible obs: 92 % / Rmerge(I) obs: 0.052 |
| Reflection shell | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 1.84 Å / Rmerge(I) obs: 0.4 / Mean I/σ(I) obs: 1.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: molecular replacement Starting model: 1OBP Resolution: 1.8→9.95 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 1113369.18 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 73.09 Å2 / ksol: 0.362 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→9.95 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.91 Å / Rfactor Rfree error: 0.023 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 10 Å / Num. reflection obs: 15530 / % reflection Rfree: 4.8 % / Rfactor obs: 0.21 / Rfactor Rfree: 0.237 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 34.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.325 / % reflection Rfree: 4.9 % / Rfactor Rwork: 0.289 |
