 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1h4t: Prolyl-tRNA synthetase from Thermus thermophilus complexed with L... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h4t | ||||||
|---|---|---|---|---|---|---|---|
| Title | Prolyl-tRNA synthetase from Thermus thermophilus complexed with L-proline | ||||||
 Components Components | PROLYL-TRNA SYNTHETASE | ||||||
 Keywords Keywords | AMINOACYL-TRNA SYNTHETASE / ATP + L-PROLINE + TRNA(PRO) AMP + PPI + L-PROLYL-TRNA(PRO) / CLASS II AMINOACYL-TRNA SYNTHETASE | ||||||
| Function / homology |  Function and homology information Function and homology informationproline-tRNA ligase / proline-tRNA ligase activity / prolyl-tRNA aminoacylation / aminoacyl-tRNA synthetase multienzyme complex / ATP binding / metal ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |   THERMUS THERMOPHILUS (bacteria) THERMUS THERMOPHILUS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Yaremchuk, A. / Tukalo, M. / Cusack, S. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2001 Title: A Succession of Substrate Induced Conformational Changes Ensures the Amino Acid Specificity of Thermus Thermophilus Prolyl-tRNA Synthetase: Comparison with Histidyl-tRNA Synthetase Authors: Yaremchuk, A. / Tukalo, M. / Grotli, M. / Cusack, S. #1: Journal: Embo J. / Year: 2000 Title: Crystal Structure of a Eukaryote/Archaeon-Like Prolyl-tRNA Synthetase and its Complex with tRNA (Pro)(Cgg) Authors: Yaremchuk, A. / Cusack, S. / Tukalo, M. #2: Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Crystallisation and Preliminary X-Ray Diffraction Analysis of Thermus Thermophilus Prolyl-tRNA Synthetase Authors: Yaremchuk, A. / Cusack, S. / Tukalo, M. #3: Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Improved Crystals of Thermus Thermophilus Prolyl-tRNA Synthetase Complexed with Cognate tRNA Obtained by Crystallisation from Precipitate Authors: Yaremchuk, A. / Krikliviy, I. / Cusack, S. / Tukalo, M. #4: Journal: Structure / Year: 1997 Title: TRNA(Pro) Recognition by Thermus Thermophilus Prolyl-tRNA Synthetase Authors: Cusack, S. / Yaremchuk, A. / Krikliviy, I. / Tukalo, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h4t.cif.gz | 381.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h4t.ent.gz | 312.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1h4t.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h4/1h4tftp://data.pdbj.org/pub/pdb/validation_reports/h4/1h4t | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1h4qC  1h4sC  1h4vC  1hc7SC C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||
| 2 | 
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 54562.965 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Details: PURIFICATION DESCRIBED IN REFERENCE 2 / Source: (natural) THERMUS THERMOPHILUS (bacteria) / Strain: HB-8 / References: UniProt: Q5SM28*PLUS, proline-tRNA ligase#2: Chemical | ChemComp-PRO / 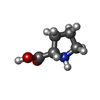  Type: L-peptide linking / Mass: 115.130 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C5H9NO2 Type: L-peptide linking / Mass: 115.130 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C5H9NO2#3: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 440 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 440 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.68 Å3/Da / Density % sol: 65 % |
|---|---|
| Crystal grow | pH: 7.9 / Details: SEE REFERENCE 2, pH 7.90 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM1A / Wavelength: 0.873 / Beamline: BM1A / Wavelength: 0.873 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Dec 15, 1996 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.873 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→15 Å / Num. obs: 71108 / % possible obs: 99 % / Redundancy: 4 % / Rmerge(I) obs: 0.068 |
| Reflection shell | Resolution: 2.9→2.97 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.223 / Mean I/σ(I) obs: 3.3 / % possible all: 99.9 |
| Reflection | *PLUS Lowest resolution: 15 Å / Num. measured all: 284962 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1HC7 (LIGAND FREE PROLYL-TRNA SYNTHETASE) Resolution: 2.9→15 Å / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: ZINC CO-ORDINATION TO FOUR CYSTEINES WAS RESTRAINED. SIDE-CHAINS ATOMS WITH OCCUPANCY ZERO HAVE POOR OR ABSENT ELECTRON DENSITY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 37.4 Å2 / ksol: 0.325 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 15 Å / Rfactor obs: 0.2 / Rfactor Rwork: 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
