Entry Database : PDB / ID : 1h0aTitle Epsin ENTH bound to Ins(1,4,5)P3 EPSIN Keywords / / / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species RATTUS NORVEGICUS (Norway rat)Method / / / Resolution : 1.7 Å Authors Ford, M.G.J. / McMahon, H.T. / Evans, P.R. Journal : Nature / Year : 2002Title : Curvature of Clathrin-Coated Pits Driven by EpsinAuthors : Ford, M.G.J. / Mills, I. / Peter, B. / Vallis, Y. / Praefcke, G. / Evans, P.R. / Mcmahon, H.T. History Deposition Jun 12, 2002 Deposition site / Processing site Revision 1.0 Oct 3, 2002 Provider / Type Revision 1.1 May 8, 2011 Group Revision 1.2 Jul 13, 2011 Group Revision 1.3 Dec 13, 2023 Group Data collection / Database references ... Data collection / Database references / Other / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



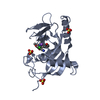





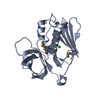

 PDBj
PDBj



 Assembly
Assembly

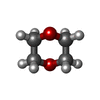
 Mass: 88.105 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H8O2
Mass: 88.105 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H8O2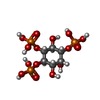
 Mass: 420.096 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15O15P3
Mass: 420.096 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15O15P3 Mass: 18.015 Da / Num. of mol.: 204 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 204 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.98
/ Beamline: ID29 / Wavelength: 0.98  Processing
Processing