 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gzu: Crystal Structure of Human Nicotinamide Mononucleotide Adenylyltr... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gzu | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Human Nicotinamide Mononucleotide Adenylyltransferase in Complex with NMN | |||||||||
 Components Components | NICOTINAMIDE MONONUCLEOTIDE ADENYLYL TRANSFERASE | |||||||||
 Keywords Keywords | TRANSFERASE / NAD BIOSYNTHESIS / ADENYLYLTRANSFERASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationprotein ADP-ribosyltransferase-substrate adaptor activity / nicotinamide-nucleotide adenylyltransferase / nicotinate-nucleotide adenylyltransferase / nicotinamide-nucleotide adenylyltransferase activity / nucleotide biosynthetic process / nicotinate-nucleotide adenylyltransferase activity / NAD+ biosynthetic process via the salvage pathway / ATP generation from poly-ADP-D-ribose / Nicotinate metabolism / nuclear body ...protein ADP-ribosyltransferase-substrate adaptor activity / nicotinamide-nucleotide adenylyltransferase / nicotinate-nucleotide adenylyltransferase / nicotinamide-nucleotide adenylyltransferase activity / nucleotide biosynthetic process / nicotinate-nucleotide adenylyltransferase activity / NAD+ biosynthetic process via the salvage pathway / ATP generation from poly-ADP-D-ribose / Nicotinate metabolism / nuclear body / negative regulation of DNA-templated transcription / chromatin / nucleoplasm / ATP binding / identical protein binding / nucleus Similarity search - Function | |||||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.9 Å | |||||||||
 Authors Authors | Werner, E. / Ziegler, M. / Lerner, F. / Schweiger, M. / Heinemann, U. | |||||||||
 Citation Citation | Journal: FEBS Lett. / Year: 2002 Title: Crystal structure of human nicotinamide mononucleotide adenylyltransferase in complex with NMN. Authors: Werner, E. / Ziegler, M. / Lerner, F. / Schweiger, M. / Heinemann, U. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gzu.cif.gz | 151.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gzu.ent.gz | 120 KB | Display | PDB format |
| PDBx/mmJSON format | 1gzu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gz/1gzuftp://data.pdbj.org/pub/pdb/validation_reports/gz/1gzu | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Beg auth comp-ID: LYS / Beg label comp-ID: LYS / End auth comp-ID: LEU / End label comp-ID: LEU / Refine code: 3 / Auth seq-ID: 6 - 270 / Label seq-ID: 17 - 281
NCS oper:
|
-Components
| #1: Protein | Mass: 33444.406 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PQE30 / Production host:  References: UniProt: Q9HAN9, nicotinamide-nucleotide adenylyltransferase #2: Chemical | 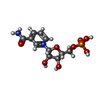  Mass: 335.227 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H16N2O8P Mass: 335.227 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H16N2O8PHas protein modification | Y | Sequence details | N-TERMINUS INCLUDES A HIS-TAG | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.71 Å3/Da / Density % sol: 66.56 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 Details: 2.0 M NA/KH2PO4, 100 M M TRIS PH 7.5, 5% 2-PROPANOL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 293 K / Method: vapor diffusion, hanging drop / Details: Werner, E., (2002) Acta Crystallogr., D58, 140. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.9795 / Wavelength: 0.9795 Å / Beamline: ID14-4 / Wavelength: 0.9795 / Wavelength: 0.9795 Å |
| Detector | Detector: CCD / Date: Jun 17, 2001 / Details: MIRRORS |
| Radiation | Monochromator: SI / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→20 Å / Num. obs: 62691 / % possible obs: 98.3 % / Observed criterion σ(I): 0.5 / Redundancy: 10.1 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 11.2 |
| Reflection shell | Resolution: 2.9→3 Å / Rmerge(I) obs: 0.492 / Mean I/σ(I) obs: 1.1 / % possible all: 92.5 |
| Reflection | *PLUS Lowest resolution: 20 Å / Num. measured all: 632186 / Rmerge(I) obs: 0.07 |
| Reflection shell | *PLUS % possible obs: 92.5 % / Num. unique obs: 5937 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.9→20 Å / Cor.coef. Fo:Fc: 0.888 / Cor.coef. Fo:Fc free: 0.85 / SU B: 8.754 / SU ML: 0.167 / Cross valid method: THROUGHOUT / ESU R: 0.801 / ESU R Free: 0.4 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|