 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3q35: Structure of the Rtt109-AcCoA/Vps75 complex and implications for ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3q35 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the Rtt109-AcCoA/Vps75 complex and implications for chaperone-mediated histone acetylation | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE/CHAPERONE / Rtt109:Vps75=2:2 stoichiometry complex / Acetyl Coenzyme A (ACoA) bound / autoacetylation at Rtt109 Lys290 / nuclear / TRANSFERASE-CHAPERONE complex | ||||||
| Function / homology |  Function and homology information Function and homology informationhistone H3K23 acetyltransferase activity / histone H3K56 acetyltransferase activity / H3 histone acetyltransferase complex / histone H3K14 acetyltransferase activity / histone H3K9 acetyltransferase activity / regulation of double-strand break repair via nonhomologous end joining / maintenance of rDNA / transposable element silencing / histone H3 acetyltransferase activity / acetyltransferase activator activity ...histone H3K23 acetyltransferase activity / histone H3K56 acetyltransferase activity / H3 histone acetyltransferase complex / histone H3K14 acetyltransferase activity / histone H3K9 acetyltransferase activity / regulation of double-strand break repair via nonhomologous end joining / maintenance of rDNA / transposable element silencing / histone H3 acetyltransferase activity / acetyltransferase activator activity / DNA replication-dependent chromatin assembly / replication-born double-strand break repair via sister chromatid exchange / histone H3K27 acetyltransferase activity / cellular response to stress / protein-lysine-acetyltransferase activity / histone acetyltransferase / double-strand break repair via nonhomologous end joining / protein transport / nucleosome assembly / chromatin organization / regulation of gene expression / histone binding / chromatin remodeling / chromatin binding / DNA damage response / regulation of transcription by RNA polymerase II / chromatin / identical protein binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.3 Å | ||||||
 Authors Authors | Tang, Y. / Yuan, H. / Meeth, K. / Marmorstein, R. | ||||||
 Citation Citation | Journal: Structure / Year: 2011 Title: Structure of the Rtt109-AcCoA/Vps75 Complex and Implications for Chaperone-Mediated Histone Acetylation. Authors: Tang, Y. / Holbert, M.A. / Delgoshaie, N. / Wurtele, H. / Guillemette, B. / Meeth, K. / Yuan, H. / Drogaris, P. / Lee, E.H. / Durette, C. / Thibault, P. / Verreault, A. / Cole, P.A. / Marmorstein, R. #1: Journal: Nat.Struct.Mol.Biol. / Year: 2008Title: Fungal Rtt109 histone acetyltransferase is an unexpected structural homolog of metazoan p300/CBP. Authors: Tang, Y. / Holbert, M.A. / Wurtele, H. / Meeth, K. / Rocha, W. / Gharib, M. / Jiang, E. / Thibault, P. / Verreault, A. / Verrault, A. / Cole, P.A. / Marmorstein, R. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 2008Title: Structure of Vps75 and implications for histone chaperone function. Authors: Tang, Y. / Meeth, K. / Jiang, E. / Luo, C. / Marmorstein, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3q35.cif.gz | 270 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3q35.ent.gz | 217.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3q35.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q3/3q35ftp://data.pdbj.org/pub/pdb/validation_reports/q3/3q35 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3q33SC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 50348.961 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: BamHI/XhoI insert Source: (gene. exp.) Gene: KIM2, L1377, REM50, RTT109, YLL002W / Plasmid: 6His-TEV-pCDF-Duet / Production host:  |
|---|---|
| #2: Protein | Mass: 27292.619 Da / Num. of mol.: 1 / Fragment: unp residues 1-232 Source method: isolated from a genetically manipulated source Details: BamHI/XhoI insert Source: (gene. exp.) Gene: N0890, VPS75, YNL246W / Plasmid: GST-TEV-pRSF-Duet / Production host: |
| #3: Chemical | ChemComp-ACO / 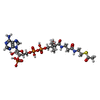  Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S |
| #4: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 11 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 11 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.02 Å3/Da / Density % sol: 59.24 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 10.0% (v/v) PEG8000; 8% (v/v) ethylene glycol; 100 mM Hepes, VAPOR DIFFUSION, HANGING DROP, temperature 298K, pH 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 23-ID-B / Wavelength: 0.988 Å / Beamline: 23-ID-B / Wavelength: 0.988 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Dec 14, 2008 / Details: mirrors |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.988 Å / Relative weight: 1 |
| Reflection | Resolution: 3.3→50 Å / Num. all: 15348 / Num. obs: 15302 / % possible obs: 99.7 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 7.1 % / Rsym value: 0.083 / Net I/σ(I): 16.5 |
| Reflection shell | Resolution: 3.3→3.42 Å / Redundancy: 6.2 % / Mean I/σ(I) obs: 3.6 / Rsym value: 0.427 / % possible all: 97.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 3Q33 Resolution: 3.3→48.01 Å / SU ML: 0.35 / Isotropic thermal model: Isotropic / σ(F): 1.34 / Phase error: 25.69 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.83 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 54.394 Å2 / ksol: 0.314 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.3→48.01 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
