 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gzm | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of Bovine Rhodopsin in a Trigonal Crystal Form | |||||||||
 Components Components | RHODOPSIN | |||||||||
 Keywords Keywords | SIGNALING PROTEIN / PHOTORECEPTOR / RETINAL PROTEIN / VISUAL PIGMENT / G-PROTEIN COUPLED RECEPTOR / INTEGRAL MEMBRANE PROTEIN / PALMITATE / PHOSPHORYLATION | |||||||||
| Function / homology |  Function and homology information Function and homology informationOpsins / VxPx cargo-targeting to cilium / rod bipolar cell differentiation / sperm head plasma membrane / opsin binding / The canonical retinoid cycle in rods (twilight vision) / absorption of visible light / G protein-coupled opsin signaling pathway / 11-cis retinal binding / podosome assembly ...Opsins / VxPx cargo-targeting to cilium / rod bipolar cell differentiation / sperm head plasma membrane / opsin binding / The canonical retinoid cycle in rods (twilight vision) / absorption of visible light / G protein-coupled opsin signaling pathway / 11-cis retinal binding / podosome assembly / G protein-coupled photoreceptor activity / photoreceptor inner segment membrane / rod photoreceptor outer segment / cellular response to light stimulus / G protein-coupled receptor complex / Inactivation, recovery and regulation of the phototransduction cascade / thermotaxis / Activation of the phototransduction cascade / detection of temperature stimulus involved in thermoception / outer membrane / response to light intensity / photoreceptor cell maintenance / arrestin family protein binding / photoreceptor outer segment membrane / G alpha (i) signalling events / phototransduction, visible light / phototransduction / response to light stimulus / photoreceptor outer segment / G-protein alpha-subunit binding / visual perception / guanyl-nucleotide exchange factor activity / microtubule cytoskeleton organization / cell-cell junction / photoreceptor disc membrane / sperm midpiece / gene expression / G protein-coupled receptor signaling pathway / Golgi membrane / zinc ion binding / membrane / identical protein binding / plasma membrane Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å | |||||||||
 Authors Authors | Li, J. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2004 Title: Structure of Bovine Rhodopsin in a Trigonal Crystal Form Authors: Li, J. / Edwards, P. / Burghammer, M. / Villa, C. / Schertler, G.F.X. #1: Journal: J.Mol.Biol. / Year: 2004 Title: Crystals of Native and Modified Bovine Rhodopsins and Their Heavy Atom Derivatives Authors: Edwards, P. / Li, J. / Burghammer, M. / Mcdowell, J.H. / Villa, C. / Hargrave, P.A. / Schertler, G.F.X. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gzm.cif.gz | 159.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gzm.ent.gz | 124.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1gzm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gz/1gzmftp://data.pdbj.org/pub/pdb/validation_reports/gz/1gzm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1f88S S: Starting model for refinement |
|---|---|
| Similar structure data |





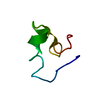











-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
-Protein / Sugars , 2 types, 6 molecules AB
| #1: Protein | Mass: 39057.492 Da / Num. of mol.: 2 / Source method: isolated from a natural source Details: DISULFIDE LINK BETWEEN A110 AND A187, AND B110 AND B187 Source: (natural) #2: Polysaccharide | beta-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta- ...beta-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source |
|---|
-Non-polymers , 7 types, 65 molecules 


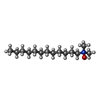



| #3: Chemical |  Mass: 284.436 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H28O Mass: 284.436 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H28O#4: Chemical | ChemComp-PLM /  Mass: 256.424 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C16H32O2 Mass: 256.424 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C16H32O2#5: Chemical |  Mass: 691.959 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C37H74NO8P / Comment: phospholipid*YM Mass: 691.959 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C37H74NO8P / Comment: phospholipid*YM#6: Chemical |  Mass: 229.402 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM Mass: 229.402 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM#7: Chemical | ChemComp-C8E / (  Mass: 306.438 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C16H34O5 / Comment: C8E, detergent*YM Mass: 306.438 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C16H34O5 / Comment: C8E, detergent*YM#8: Chemical |  Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#9: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 40 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Compound details | LIGHT-ABSORBING MOLECULES THAT MEDIATE VISION. CONSIST OF AN APOPROTEIN, OPSIN, COVALENTLY BOUND TO ...LIGHT-ABSORBING MOLECULES THAT MEDIATE VISION. CONSIST OF AN APOPROTEIN |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 4 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.07 Å3/Da / Density % sol: 49 % Description: DATA WERE COLLECTED USING MICROFOCUSED SYNCHROTRON SOURCE. | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, sitting drop / pH: 8.5 Details: VAPOUR DIFFUSION IN SITTING DROPS OF 15 MG/ML PROTEIN AND 0.2% C8E4, 0.05%LDAO AGAINST 0.8M LI2SO4, 1.6% PEG8000 AND 20% GLYCEROL, pH 8.5 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 18 ℃ / Method: vapor diffusion, sitting drop / Details: Edwards, P., (2004) J. Mol. Biol., 343, 1439. | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID13 / Wavelength: 0.782 / Beamline: ID13 / Wavelength: 0.782 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Oct 15, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.782 Å / Relative weight: 1 |
| Reflection | Resolution: 2.65→46 Å / Num. obs: 26026 / % possible obs: 97 % / Observed criterion σ(I): 0 / Redundancy: 4.4 % / Biso Wilson estimate: 58.2 Å2 / Rmerge(I) obs: 0.119 / Net I/σ(I): 11 |
| Reflection shell | Resolution: 2.65→2.79 Å / Redundancy: 1.6 % / Rmerge(I) obs: 0.434 / Mean I/σ(I) obs: 1.4 / % possible all: 86 |
| Reflection | *PLUS % possible obs: 97 % |
| Reflection shell | *PLUS % possible obs: 86.1 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1F88 Resolution: 2.65→46 Å / Rfactor Rfree error: 0.0066 / Data cutoff high absF: 10000000 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: THE FOLLOWING HOH RESIDUES ARE EQUIVALENT BY NON- CRYSTALLOGRAPHIC SYMMETRY AND ARE EQUIVALENT TO THESE RESIDUES IN THE PRIMARY REFERENCE: U1 == V1, AND WATER RESIDUE 7 IN REFERENCE U2 == ...Details: THE FOLLOWING HOH RESIDUES ARE EQUIVALENT BY NON- CRYSTALLOGRAPHIC SYMMETRY AND ARE EQUIVALENT TO THESE RESIDUES IN THE PRIMARY REFERENCE: U1 == V1, AND WATER RESIDUE 7 IN REFERENCE U2 == V3, AND WATER RESIDUE 16 IN REFERENCE U3 == V4, AND WATER RESIDUE 8 IN REFERENCE U4 == V5, AND WATER RESIDUE 11 IN REFERENCE U5 == V6, AND WATER RESIDUE 4 IN REFERENCE U6 == V7, AND WATER RESIDUE 13 IN REFERENCE U7 == V11, AND WATER RESIDUE 6 IN REFERENCE U8 == V9, AND WATER RESIDUE 3 IN REFERENCE U9 == V19, AND WATER RESIDUE 15 IN REFERENCE U10 == V3, AND WATER RESIDUE 18 IN REFERENCE U11 == V13, AND WATER RESIDUE 5 IN REFERENCE U12 == V14, AND WATER RESIDUE 14 IN REFERENCE U13 == V15, AND WATER RESIDUE 19 IN REFERENCE U14 == V16, AND WATER RESIDUE 9 IN REFERENCE U15 == V17, AND WATER RESIDUE 1 IN REFERENCE U16 == V12, AND WATER RESIDUE 17 IN REFERENCE U17 == V18, AND WATER RESIDUE 20 IN REFERENCE U18 == V2, AND WATER RESIDUE 12 IN REFERENCE U19 == V19, AND WATER RESIDUE 2 IN REFERENCE U20 == V20, AND WATER RESIDUE 10 IN REFERENCE THE FOLLOWING CARBOHYDRATE RESIDUES ARE EQUIVALENT BY NON- CRYSTALLOGRAPHIC SYMMETRY: A1335 IS EQUIVALENT TO B1335 A1336 IS EQUIVALENT TO B1336 A1337 IS EQUIVALENT TO B1350 A1338 IS EQUIVALENT TO B1337 A1339 IS EQUIVALENT TO B1338 A1340 IS EQUIVALENT TO B1339
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 56 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.65→46 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Rms dev Biso : 1 Å2 / Rms dev position: 0.515 Å / Weight position: 5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.65→2.74 Å / Rfactor Rfree error: 0.0275 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 46 Å / % reflection Rfree: 5 % / Rfactor Rfree: 0.235 / Rfactor Rwork: 0.202 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.315 / Rfactor Rwork: 0.312 |
