 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gtv: CRYSTAL STRUCTURE OF MYCOBACTERIUM TUBERCULOSIS THYMIDYLATE KINAS... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gtv | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF MYCOBACTERIUM TUBERCULOSIS THYMIDYLATE KINASE COMPLEXED WITH THYMIDINE-5'-DIPHOSPHATE (TDP) | |||||||||
 Components Components | THYMIDYLATE KINASE | |||||||||
 Keywords Keywords | TRANSFERASE / TRANSFERASE (ATP:TMP PHOSPHOTRANSFERASE) / KINASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationTMP metabolic process / dTMP kinase / dUDP biosynthetic process / dTDP biosynthetic process / dTMP kinase activity / dTTP biosynthetic process / GTP binding / magnesium ion binding / protein homodimerization activity / ATP binding ...TMP metabolic process / dTMP kinase / dUDP biosynthetic process / dTDP biosynthetic process / dTMP kinase activity / dTTP biosynthetic process / GTP binding / magnesium ion binding / protein homodimerization activity / ATP binding / metal ion binding / cytosol / cytoplasm Similarity search - Function | |||||||||
| Biological species |   MYCOBACTERIUM TUBERCULOSIS (bacteria) MYCOBACTERIUM TUBERCULOSIS (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.55 Å X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.55 Å | |||||||||
 Authors Authors | Ursby, T. / Weik, M. / Fioravanti, E. / Delarue, M. / Goeldner, M. / Bourgeois, D. | |||||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2002 Title: Cryophotolysis of Caged Compounds: A Technique for Trapping Intermediate States in Protein Crystals Authors: Ursby, T. / Weik, M. / Fioravanti, E. / Delarue, M. / Goeldner, M. / Bourgeois, D. #1: Journal: J.Mol.Biol. / Year: 2001Title: X-Ray Structure of Tmp Kinase from Mycobacterium Tuberculosis Complexed with Tmp at 1.95 A Resolution Authors: De La Sierra Li, I. / Munier-Lehmann, H. / Gilles, A.M. / Barzu, O. / Delarue, M. #2: Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Crystallization and Preliminary X-Ray Analysis of the Thymidylate Kinase from Mycobacterium Tuberculosis Authors: De La Sierra Li, I. / Munier-Lehmann, H. / Gilles, A.M. / Barzu, O. / Delarue, M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gtv.cif.gz | 106.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gtv.ent.gz | 81 KB | Display | PDB format |
| PDBx/mmJSON format | 1gtv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gt/1gtvftp://data.pdbj.org/pub/pdb/validation_reports/gt/1gtv | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
| ||||||||
| Details | NOTE THAT THE CRYSTAL CONTAINED 48% OF THE COMPLEX WITH TDP(TYD) AND 52% OF THE COMPLEX WITH TMP, WHICH IS REFLECTEDIN THE TWO MODELS IN THE STRUCTURE FILE. THE MODEL OF THECOMPLEX WITH TDP (TYD) IS OBTAINED BY TAKING CHAIN A, INCREASING THE OCCUPANCY BY A FACTOR 1/0.48 AND ADDING THEWATER MOLECULES WITH OCCUPANCY 1. |
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 22662.525 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) MYCOBACTERIUM TUBERCULOSIS (bacteria) / Plasmid: PET22B / Production host: References: UniProt: O05891, UniProt: P9WKE1*PLUS, dTMP kinase |
|---|
-Non-polymers , 6 types, 300 molecules 
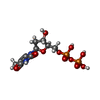


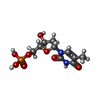

| #2: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-TYD / |  Mass: 402.188 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N2O11P2 Mass: 402.188 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N2O11P2#4: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4#5: Chemical | ChemComp-ACT /  Mass: 59.044 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H3O2#6: Chemical | ChemComp-TMP / |  Mass: 322.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O8P Mass: 322.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O8P#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 284 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50 % / Description: STARTING MODEL PDB ENTRY 1GSI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6 Details: PROTEIN CRYSTALLIZED IN 1.4M AMMONIUM SULFATE,100MM MES PH6, 2% PEG 2000 25MM MAGNESIUM ACETATE,2MM BETA-MERCAPTOETHANOL, pH 6.00 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 293 K / pH: 7.4 / Method: vapor diffusion, hanging dropDetails: Li de La Sierra, I., (2000) Acta Crystallogr.,Sect., D56, 226. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.933 / Beamline: ID14-2 / Wavelength: 0.933 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jul 10, 2000 / Details: TOROIDAL MIRROR |
| Radiation | Monochromator: DIAMOND(111), GE(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 1.55→25.3 Å / Num. obs: 33359 / % possible obs: 97.5 % / Redundancy: 10.8 % / Biso Wilson estimate: 18.4 Å2 / Rmerge(I) obs: 0.065 / Net I/σ(I): 26.2 |
| Reflection shell | Resolution: 1.55→1.63 Å / Redundancy: 6.7 % / Rmerge(I) obs: 0.179 / Mean I/σ(I) obs: 9 / % possible all: 96 |
| Reflection | *PLUS Num. measured all: 360419 |
| Reflection shell | *PLUS % possible obs: 96 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 1.55→25.27 Å / Rfactor Rfree error: 0.005 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: RESIDUES 36-46, 73-81, 94 AND 159- 166 WERE MODELED IN A TYD CONFORMATION (CHAIN A), WHICH WAS REFINED, AND A TMP CONFORMATION (CHAIN B) TAKEN FROM PDB ENTRY 1GSI AND KEPT FIXED. THE REST OF ...Details: RESIDUES 36-46, 73-81, 94 AND 159- 166 WERE MODELED IN A TYD CONFORMATION (CHAIN A), WHICH WAS REFINED, AND A TMP CONFORMATION (CHAIN B) TAKEN FROM PDB ENTRY 1GSI AND KEPT FIXED. THE REST OF PROTEIN CHAINS A AND B WERE REFINED AND IDENTICAL FOR CHAINS A AND B. HET ATOMS AND WATER MOLECULES ANNOTATED AS CHAIN A OR B CORRESPOND TO THE TYD AND THE TMP CONFORMATIONS RESPECTIVELY, WITH THE TMP CONFORMATION ATOMS KEPT FIXED DURING REFINEMENT TO THE 1GSI COORDINATES.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 57.4639 Å2 / ksol: 0.353042 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.55→25.27 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.55→1.65 Å / Rfactor Rfree error: 0.015 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.206 |
