 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gl6: Plasmid coupling protein TrwB in complex with the non-hydrolysabl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gl6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Plasmid coupling protein TrwB in complex with the non-hydrolysable GTP analogue GDPNP | ||||||
 Components Components | ATPASE | ||||||
 Keywords Keywords | COUPLING PROTEIN / TYPE IV SECRETION SYSTEM / BACTERIAL CONJUG PROTEIN / RING HELICASE | ||||||
| Function / homology |  Function and homology information Function and homology informationvesicle-fusing ATPase / nucleotide binding / metal ion binding / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Gomis-Ruth, F.X. / Moncalian, G. / De La cruz, F. / Coll, M. | ||||||
 Citation Citation | Journal: Nature / Year: 2001 Title: The Bacterial Conjugation Protein Trwb Resembles Ring Helicases and F1-ATPase Authors: Gomis-Ruth, F.X. / Moncalian, G. / Perez-Luque, R. / Gonzalez, A. / Cabezon, E. / De La Cruz, F. / Coll, M. #1: Journal: J.Biol.Chem. / Year: 2002 Title: Conjugative Plasmid Protein Trwb, an Integral Membrane Type Iv Secretion System Coupling Protein. Detailed Structural Features and Mapping of the Active Site Cleft. Authors: Gomis-Ruth, F.X. / Moncalian, G. / De La Cruz, F. / Coll, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gl6.cif.gz | 500.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gl6.ent.gz | 411.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1gl6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gl/1gl6ftp://data.pdbj.org/pub/pdb/validation_reports/gl/1gl6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1e9rSC  1e9sC  1gkiC  1gl7C S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj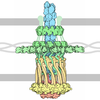





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 48558.312 Da / Num. of mol.: 6 / Fragment: CYTOSOLIC FRAGMENT / Mutation: YES Source method: isolated from a genetically manipulated source Details: COMPLEX WITH GDPNP. / Source: (gene. exp.) #2: Chemical | ChemComp-GNP / 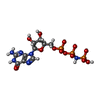  Mass: 522.196 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C10H17N6O13P3 Mass: 522.196 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C10H17N6O13P3Comment: GppNHp, GMPPNP, energy-carrying molecule analogue*YM #3: Chemical | ChemComp-CL / |   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-EPE / |   Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 589 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 589 / Source method: isolated from a natural source / Formula: H2OCompound details | CHAIN A, B, D, E, F, G FIRST 70 RESIDUES DELETED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 57 % |
|---|---|
| Crystal grow | pH: 7.5 / Details: pH 7.50 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.842 / Beamline: ID14-2 / Wavelength: 0.842 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.842 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→50 Å / Num. obs: 81160 / % possible obs: 98 % / Redundancy: 5.3 % / Biso Wilson estimate: 76.7 Å2 / Rmerge(I) obs: 0.093 / Net I/σ(I): 6.1 |
| Reflection shell | Resolution: 2.8→2.95 Å / Redundancy: 5.3 % / Rmerge(I) obs: 0.453 / Mean I/σ(I) obs: 1.7 / % possible all: 92 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1E9R Resolution: 2.8→50 Å / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: FREE RFACTOR USED UNTIL PENULTIMATE REFINEMENT CYCLE. SAME INDICES AS IN 1E9R, 1GKI AND 1GL7.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 56 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
