 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gil: STRUCTURE OF ACTIVE CONFORMATIONS OF GIA1 AND THE MECHANISM OF GT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gil | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF ACTIVE CONFORMATIONS OF GIA1 AND THE MECHANISM OF GTP HYDROLYSIS | ||||||
 Components Components | G PROTEIN GI ALPHA 1 | ||||||
 Keywords Keywords | GTP-BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationadenylate cyclase regulator activity / Extra-nuclear estrogen signaling / Adenylate cyclase inhibitory pathway / negative regulation of synaptic transmission / Adrenaline,noradrenaline inhibits insulin secretion / ADP signalling through P2Y purinoceptor 12 / GTPase activating protein binding / G alpha (i) signalling events / neurotransmitter receptor localization to postsynaptic specialization membrane / negative regulation of insulin secretion ...adenylate cyclase regulator activity / Extra-nuclear estrogen signaling / Adenylate cyclase inhibitory pathway / negative regulation of synaptic transmission / Adrenaline,noradrenaline inhibits insulin secretion / ADP signalling through P2Y purinoceptor 12 / GTPase activating protein binding / G alpha (i) signalling events / neurotransmitter receptor localization to postsynaptic specialization membrane / negative regulation of insulin secretion / centriolar satellite / adenylate cyclase inhibitor activity / positive regulation of protein localization to cell cortex / T cell migration / D2 dopamine receptor binding / adenylate cyclase-inhibiting serotonin receptor signaling pathway / G protein-coupled serotonin receptor binding / cellular response to forskolin / regulation of mitotic spindle organization / chemokine-mediated signaling pathway / response to prostaglandin E / positive regulation of cholesterol biosynthetic process / G protein-coupled receptor binding / G-protein beta/gamma-subunit complex binding / adenylate cyclase-modulating G protein-coupled receptor signaling pathway / adenylate cyclase-inhibiting G protein-coupled receptor signaling pathway / GDP binding / heterotrimeric G-protein complex / G protein activity / midbody / cell cortex / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / postsynapse / ciliary basal body / G protein-coupled receptor signaling pathway / cell division / GTPase activity / centrosome / nucleolus / GTP binding / glutamatergic synapse / Golgi apparatus / magnesium ion binding / protein-containing complex / nucleoplasm / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.3 Å X-RAY DIFFRACTION / Resolution: 2.3 Å | ||||||
 Authors Authors | Coleman, D.E. / Berghuis, A.M. / Sprang, S.R. | ||||||
 Citation Citation | Journal: Science / Year: 1994 Title: Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Authors: Coleman, D.E. / Berghuis, A.M. / Lee, E. / Linder, M.E. / Gilman, A.G. / Sprang, S.R. #1: Journal: J.Mol.Biol. / Year: 1994Title: Crystallization and Preliminary Crystallographic Studies of Gia1 and Mutants of Gia1 in the GTP and Gdp-Bound States Authors: Coleman, D.E. / Lee, E. / Mixon, M.B. / Linder, M.E. / Berghuis, A.M. / Gilman, A.G. / Sprang, S.R. | ||||||
| History |
| ||||||
| Remark 650 | HELIX HELIX HELIX_ID: AA,DISTORTED AT GLN 68 AND ILE 85. HELIX_ID: AB,BOTH TERMINI ARE IN 3_10 ...HELIX HELIX HELIX_ID: AA,DISTORTED AT GLN 68 AND ILE 85. HELIX_ID: AB,BOTH TERMINI ARE IN 3_10 HELIX CONFORMATION. HELIX_ID: AD,FROM ALA 138 ONWARDS HELIX IS IN 3/10 CONFORMATION. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gil.cif.gz | 76.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gil.ent.gz | 56.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1gil.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gi/1gilftp://data.pdbj.org/pub/pdb/validation_reports/gi/1gil | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 40252.863 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #3: Chemical | ChemComp-GSP / 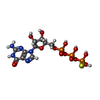  Mass: 539.246 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3S Mass: 539.246 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3S |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O |
| Nonpolymer details | COMPND GTP-GAMMA-S IS NON-HYDROLYZAB |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.48 Å3/Da / Density % sol: 50.32 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 21 ℃ / pH: 6 / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | Num. obs: 16167 / % possible obs: 97.1 % / Observed criterion σ(I): 0 |
| Reflection | *PLUS Highest resolution: 2.3 Å / Lowest resolution: 15 Å / % possible obs: 97.2 % / Redundancy: 3.3 % / Num. measured all: 58697 / Rmerge(I) obs: 0.041 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.3→8 Å / σ(F): 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.3 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 8 Å / Rfactor obs: 0.23 / Rfactor Rfree: 0.31 / Rfactor Rwork: 0.23 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_deg / Dev ideal: 1.588 |
