 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1g5u | ||||||
|---|---|---|---|---|---|---|---|
| Title | LATEX PROFILIN HEVB8 | ||||||
 Components Components | PROFILIN | ||||||
 Keywords Keywords | ALLERGEN / ACTIN-BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |   Hevea brasiliensis (rubber tree) Hevea brasiliensis (rubber tree) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.1 Å | ||||||
 Authors Authors | Fedorov, A.A. / Fedorov, E.V. / Ganglberger, E. / Breiteneder, H. / Almo, S.C. | ||||||
 Citation Citation | Journal: To be Published Title: A Comparative Structural Analysis of Allergen Profilins HEVB8 and BETV2 Authors: Fedorov, A.A. / Fedorov, E.V. / Ganglberger, E. / Breiteneder, H. / Almo, S.C. #1: Journal: INT.ARCH.ALLERGY.IMMUNOL / Year: 2000Title: Latex Allergy in Children Authors: Niggemann, B. / Breiteneder, H. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 1994Title: X-ray Structures of Isoforms of the Actin-binding Protein Profilin that Differ in their Affinity for Phosphatidylinositol Phosphates Authors: Fedorov, A.A. / Magnus, K.A. / Graupe, M.H. / Lattman, E.E. / Pollard, T.D. / Almo, S.C. #3: Journal: J.Mol.Biol. / Year: 1994Title: PURIFICATION, CHARACTERIZATION AND CRYSTALLIZATION OF HUMAN PLATELET PROFILIN EXPRESSED IN ESCHERICHIA COLI Authors: FEDOROV, A.A. / POLLARD, T.D. / ALMO, S.C. #4: Journal: Structure / Year: 1997Title: The Molecular Basis for Allergen Cross-reactivity: Crystal Structure and IgE-epitope Mapping of Birch Pollen Profilin Authors: FEDOROV, A.A. / BALL, T. / M MAHONEY, N. / VALENTA, R. / ALMO, S.C. #5: Journal: J.STRUCT.BIOL. / Year: 1998Title: CRYSTAL PACKING INDUCES A COnFORMATIONAL CHANGE IN PROFILIN-I FROM ACANTHAMOEBA CASTELLANII Authors: LIU, S. / FEDOROV, A.A. / POLLARD, T.D. / LATTMAN, E.E. / ALMO, S.C. / A MAGNUS, K. #6: Journal: Nat.Struct.Biol. / Year: 1999Title: PROFILIN BINDS PROLINE-RICH LIGANDS IN TWO DISTINCT AMIDE BACKBONE ORIENTATIONS Authors: Mahoney, N.M. / Rozwarski, D.A. / Fedorov, E. / Fedorov, A.A. / Almo, S.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1g5u.cif.gz | 62 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1g5u.ent.gz | 45.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1g5u.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g5/1g5uftp://data.pdbj.org/pub/pdb/validation_reports/g5/1g5u | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1a0kS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 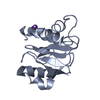
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a monomer generated from the chain A or from the chain B |
-Components
| #1: Protein | Mass: 14048.929 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Hevea brasiliensis (rubber tree) / Production host:  #2: Chemical | ChemComp-NA / |   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 57.6 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop / pH: 6 Details: sodium citrate, sucrose , pH 6.0, VAPOR DIFFUSION, HANGING DROP, temperature 290.0K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X9B / Wavelength: 0.98 Å / Beamline: X9B / Wavelength: 0.98 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Sep 22, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→20 Å / Num. all: 7618 / Num. obs: 7618 / % possible obs: 98.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.4 % / Rmerge(I) obs: 0.045 / Net I/σ(I): 18.3 |
| Reflection shell | Resolution: 2.8→2.9 Å / Redundancy: 1.8 % / Rmerge(I) obs: 0.16 / Mean I/σ(I) obs: 4.8 / % possible all: 98.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1A0K Resolution: 3.1→19.3 Å / Rfactor Rfree error: 0.013 / Data cutoff high absF: 10000 / Isotropic thermal model: restrained / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: flat model / Bsol: 43.85 Å2 / ksol: 0.242 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 41.8 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.1→19.3 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: restraints | ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
