 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
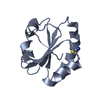
Yorodumi- PDB-1ep7: CRYSTAL STRUCTURE OF WT THIOREDOXIN H FROM CHLAMYDOMONAS REINHARDTII -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ep7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF WT THIOREDOXIN H FROM CHLAMYDOMONAS REINHARDTII | ||||||
 Components Components | THIOREDOXIN CH1, H-TYPE | ||||||
 Keywords Keywords | ELECTRON TRANSPORT | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |   Chlamydomonas reinhardtii (plant) Chlamydomonas reinhardtii (plant) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.1 Å X-RAY DIFFRACTION / Resolution: 2.1 Å | ||||||
 Authors Authors | Menchise, V. / Corbier, C. / Didierjean, C. / Saviano, M. / Benedetti, E. / Jacquot, J.P. / Aubry, A. | ||||||
 Citation Citation | Journal: Biochem.J. / Year: 2001 Title: Crystal structure of the wild-type and D30A mutant thioredoxin h of Chlamydomonas reinhardtii and implications for the catalytic mechanism. Authors: Menchise, V. / Corbier, C. / Didierjean, C. / Saviano, M. / Benedetti, E. / Jacquot, J.P. / Aubry, A. #1: Journal: Eur.J.Biochem. / Year: 1997Title: NMR Solution Structure of an Oxidised Thioredoxin h from the Eukaryotic Green Alga Chlamydomonas reinhardtii Authors: Mittard, V. / Blackledge, M.J. / Stein, M. / Jacquot, J.P. / Marion, D. / Lancelin, J.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ep7.cif.gz | 51.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ep7.ent.gz | 38 KB | Display | PDB format |
| PDBx/mmJSON format | 1ep7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ep/1ep7ftp://data.pdbj.org/pub/pdb/validation_reports/ep/1ep7 | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 11727.534 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Chlamydomonas reinhardtii (plant) / Production host:  #2: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 57 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 57 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44.17 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: PEG 8000, PEG 10000, cacodylate, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR591 / Wavelength: 1.5418 |
| Detector | Type: MACSCIENCE / Detector: IMAGE PLATE / Date: Apr 20, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→20 Å / Num. all: 42095 / Num. obs: 12186 / % possible obs: 95.1 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 4 % / Rmerge(I) obs: 0.062 / Net I/σ(I): 0.301 |
| Reflection shell | Resolution: 2.1→2.17 Å / Redundancy: 3 % / Rmerge(I) obs: 0.3 / Num. unique all: 1156 / % possible all: 92.6 |
| Reflection | *PLUS Lowest resolution: 20 Å / Num. measured all: 42095 |
| Reflection shell | *PLUS % possible obs: 92.6 % / Rmerge(I) obs: 0.301 / Mean I/σ(I) obs: 3.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.1→20 Å / σ(F): 0 / σ(I): 0 Details: The two molecules, A and B, have been refined independently, without imposing non-crystallographic 2-fold axis.
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→20 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.1 Å / Lowest resolution: 20 Å / σ(F): 0 / Rfactor obs: 0.204 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
